3 Legemiddelsektoren i Norge
3.1 GENERELT OM LEGEMIDDELBRUK
3.1.1 Nasjonalt
I Norge, som i andre land, har legemiddelomsetningen økt sterkt de siste årene. Tabell 3.1 illustrerer økningen i omsetningstallene i private og offentlige apotek, fra ca. 1,5 mrd. kroner i 1980 til 7,6 mrd. kroner i 1995. Målt i faste 1995-priser (legemiddelpriser deflatert med konsumprisindeksen) tilsvarer dette en vekst på 4 025 mill. kroner. Dette innebærer en samlet realvekst på 113 pst., svarende til en årlig reell vekst på ca. 5,2 pst. i gjennomsnitt.
Tabell 3.1 Private og offentlige apoteks legemiddelomsetning, 1980-1995
| Legemiddelsalg*) (mill. kroner, løpende priser) | Legemiddelsalg**) (mill. kroner, faste 95-priser) | Legemiddelsalg pr. innb. *) (faste 95-priser, kroner) | |
| 1980 | 1.515 | 3.552 | 868 |
| 1985 | 2.701 | 4.112 | 989 |
| 1988 | 4.005 | 4.903 | 1.162 |
| 1989 | 4.350 | 5.092 | 1.203 |
| 1990 | 4.967 | 5.586 | 1.314 |
| 1991 | 5.484 | 5.963 | 1.395 |
| 1992 | 5.838 | 6.203 | 1.443 |
| 1993 | 6.418 | 6.667 | 1.542 |
| 1994 | 6.729 | 6.893 | 1.585 |
| 1995 | 7.577 | 7.577 | 1.734 |
*) Inkl. merverdiavgift
**) Deflatert med konsumprisindeksen,
Kilde: Statens helsetilsyn, SSB 1995
Den økte omsetningsverdien er et resultat av generell prisøkning, økt forbruk og overgang til nye og dyrere preparater.
Tabell 3.2 Omsetningsveksten fordelt på prisendring, volumendring og overgang til nye og dyrere preparater 1980-1994, prosent.
| År | Prisendring*) | Volum endring**) | Netto overgang til nye og dyrere prep. | Sum, total omsetningsøkn. |
| 80-81 | 2,7 | 0,1 | 4,8 | 7,6 |
| 81-82 | 9,7 | 3,1 | 5,1 | 17,9 |
| 82-83 | 4,8 | 3,4 | 7,2 | 15,4 |
| 83-84 | 4,3 | -1,3 | 5,1 | 8,1 |
| 84-85 | 3,6 | 4,6 | 4,9 | 13,1 |
| 85-86 | 5,2 | 0,9 | 4,2 | 10,3 |
| 86-87 | 4,0 | 2,0 | 9,7 | 15,7 |
| 87-88 | 7,8 | 1,0 | 6,2 | 15,0 |
| 88-89 | 2,0 | -1,0 | 7,9 | 8,9 |
| 89-90 | 2,3 | 3,7 | 6,7 | 12,7 |
| 90-91 | 5,2 | 1,6 | 6,6 | 13,4 |
| 91-92 | 2,7 | 2,4 | 5,5 | 10,6 |
| 92-93 | -1,2 | 5,3 | 5,8 | 9,9 |
| 93-94 | -2,4 | 3,8 | 6,1 | 7,5 |
| 94-95 | 0,7 | 5,0 | 6,4 | 12,1 |
*) Prisendring for de preparater som er i markedet, målt i apotekenes innkjøpspriser (AIP)
**) Definerte døgndoser
Kilde: NMD, 1995.
Tabell 3.2 viser at det er overgang til relativt dyrere legemidler som har bidratt mest til omsetningsøkningen for legemidler de seneste årene.
Legemiddelforbruk angitt i omsetningsverdi sier lite om mengden legemidler som er brukt. Norge har derfor utviklet andre måleenheter for legemiddelforbruk. Enheten definerte døgndoser (DDD) anvendes internasjonalt til presentasjon og sammenligning av legemiddelforbruk, og er definert som «den gjennomsnittlige dose pr. døgn for et voksent menneske for preparatets vanligste bruksmåte».
Figur 3.1 viser veksten i AIP-salget, og innovative legemidlers andel av totalt AIP-salg. Figur 3.2 viser hvordan veksten i legemiddelforbruket fordeles på enkelte legemiddelgrupper. Det fremgår at det nettopp er den nye legemiddelteknologi innenfor hjerte - karmedisin, nevrologi, psykiatri, magesår og astma som står for økningen. Eldre medisiner bidrar ikke til salgsveksten.
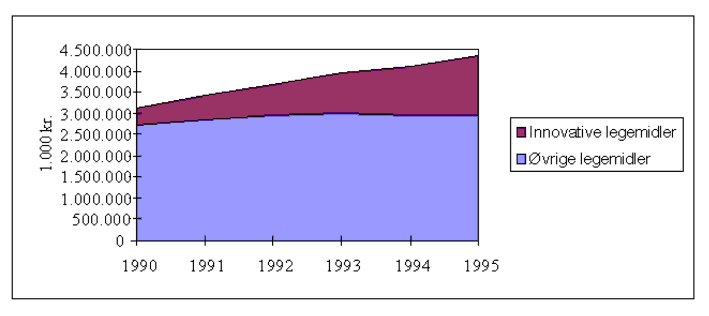
Figur 3.1 Innovative legemidlers andel av totalt AIP-salg, 1990-1995 i faste 1995-kroner (1 000 kroner).
Kilde: LMI
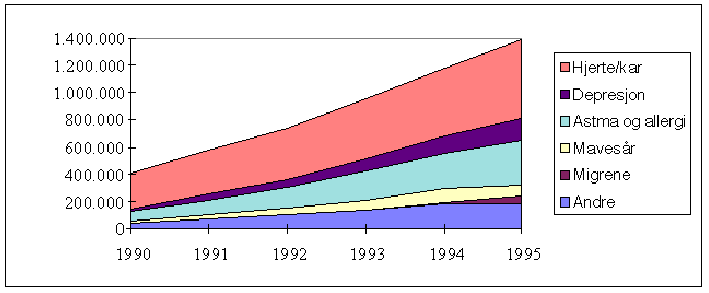
Figur 3.2 Vekst i utgifter til legemidler (AIP) fordelt på ulike diagnosegrupper (ATC-grupper). Faste 1995-kroner (1 000 kroner).
Kilde: LMI
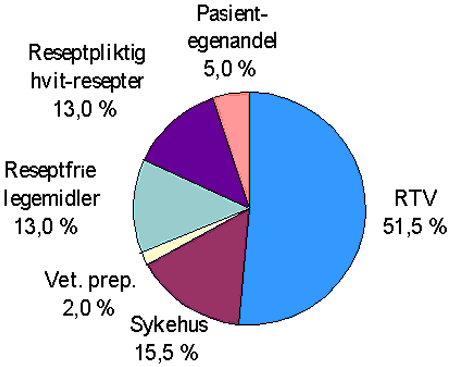
Figur 3.3 Fordelingen av legemiddelsalget i 1995
Kilde: Legemiddelindustriforeningen, 1996.
Figur 3.3 illustrerer hvem som betaler hva av det totale legemiddelsalget.
51,5 pst. av samlet salg av legemidler dekkes av folketrygden. Videre dekkes 15,5 pst. av offentlige sykehus. Til sammen dekkes 2/3 av utgiftene forbundet med legemiddelforbruket over offentlige budsjetter, mens 1/3 av utgiftene er dekket av den enkelte forbruker.
Tabell 3.3 viser utviklingen i Rikstrygdeverkets (RTVs) refusjon av utgifter og sykehusenes kjøp av legemidler i perioden 1990-95.
Tabell 3.3 RTVs refusjon av utgifter til legemidler og sykehusenes kjøp av legemidler 1990-95. Tallene i mill. kroner og løpende priser.
| RTVs utgifter til legemidler på blå resept | Pst. vekst | Sykehusenes kjøp av legemidler | Pst. vekst | |
| 1990 | 2.221 | 15,1 | 904 | 10,1 |
| 1991 | 2.561 | 15,3 | 955 | 5,6 |
| 1992 | 2.831 | 10,5 | 1.018 | 6,6 |
| 1993 | 3.035 | 7,2 | 1.079 | 5,9 |
| 1994 | 3.264 | 7,5 | 1.135 | 5,2 |
| 1995 | 3.665 | 12,3 | 1.169 | 3,0 |
Kilde: LMI og RTV, 1995-96.
Tabellen viser at gjennomsnittlig nominell årlig utgiftsvekst for refusjon av legemidler på blå resept var på 11,3 pst., dette tilsvarer en realvekst på 8 pst. Veksten i sykehusenes legemiddelutgifter var vesentlig lavere i samme periode. Myndighetene har de senere år gjennomført en rekke tiltak for å bremse utgiftsveksten for legemidler på blåresept. I løpet av 1992 ble egenandelen for legemidler hevet og et krav om at legene skal forskrive billigste synonympreparat ble innført i 1991. Videre ble det i 1993 innført et referanseprissystem (nærmere omtalt under kapittel 3.3). Tiltakene førte til lavere utgiftsvekst i 1993 og 1994 enn i de foregående årene. Den sterke veksten i 1995 er først og fremst knyttet til omsetningen av enkelte nye preparater som er tatt opp på preparatlisten bl.a. Imigran (mot migrene), Seroxat (anti depressivum) og Zocor (kolesterolsenkende).
Tabell 3.4 Legemiddelforbuket i Norge i forhold til samfunnsøkonomiske nøkkeltall
| Offentlige utgifter | Offentlige utgifter | ||||
| År | BNP | til helsevesenet | til legemidler | ||
| Pst. av | Pst. av off. utg. | ||||
| Mill. kr. | Mill. kr. | BNP | Mill. kr. | til helsevesen | |
| 1988 | 639.591 | 42.351 | 6,6 | 2.573 | 6,1 |
| 1989 | 682.347 | 43.105 | 6,3 | 2.806 | 6,5 |
| 1990 | 722.071 | 45.675 | 6,3 | 3.191 | 7,0 |
| 1991 | 762.774 | 50.352 | 6,6 | 3.582 | 7,1 |
| 1992 | 784.296 | 52.550 | 6,7 | 3.953 | 7,5 |
| 1993 | 823.339 | 54.036 | 6,6 | 4.277 | 7,9 |
| 1994 | 869.742 | 56.145 | 6,5 | 4.585 | 8,2 |
| 1995 | 925.866 | 59.513 | 6,4 | 5.053 | 8,5 |
Kilde: Statistisk sentralbyrå
Tabell 3.4 viser at offentlige utgifter til helsevesenet som andel av BNP har vært omtrent den samme i perioden 1988-95. Legemidlenes andel av ressursene til helsevesenet har vist en økende tendens. Dette indikerer at kostnadsveksten på legemiddelområdet er sterkere enn på andre områder av helsevesenet.
Legemiddelbruk i sykehus og øvrige institusjoner.
Legemiddelbruken knyttet til pasienter på sykehus finansieres over sykehusets budsjett så lenge pasienten er innlagt/behandles på sykehuset, dvs. at utgiftene til legemiddelbruken i sykehus i hovedsak dekkes av fylkeskommunene eller staten.
Når det gjelder kommunenes finansieringsansvar, ble det fra 1.1.95 gjennomført en endring i regelverket for institusjoner etter sosialtjenesteloven. Dette innebar at sykehjem, aldershjem og boliger med heldøgns omsorgstjenester dekker utgifter til helsetjenester for sine beboere (beboere betaler vederlag etter vederlagsforskriften). Tidligere dekket sykehjemmene alle utgifter over sine budsjett, mens aldershjem og boliger med heldøgns omsorgstjenester etter sosialtjenesteloven fikk dekket utgifter til legehjelp, medisiner mv. over folketrygden.
Bruk av legemidler i fylkene
Statistikk over legemiddelforbruk og forbruksmønstre viser at det er til dels store forskjeller når det gjelder bruk av legemidler fra fylke til fylke. Disse forskjellene gjelder både hvor mye legemidler som brukes totalt, og forbruket av ulike typer legemidler. Fylkesforskjellene holder seg relativt konstante fra år til år.
Den nasjonale statistikken over legemiddelforbruket, målt i omsetning pr. innbygger, viser at hver nordmann i gjennomsnitt bruker legemidler for om lag 1 500 kroner i året. Tabell 3.5 viser omsetningen av legemidler fra apotek (målt i apotekenes utsalgspris - AUP) fordelt på antall innbyggere i de enkelte fylkene. Oslo og Østfold har det høyeste forbruket og Finnmark har det laveste. Forbruket i Oslo og Akershus må ses i sammenheng pga. geografisk nærhet.
Tabell 3.5 Fylkesvis omsetning av legemidler i 1994 (apotekenes utsalgspris-AUP)
| Fylke | Omsetning pr. innbygg. (kroner) | Total omsetning (mill. kr) | Sammenlikning med lands gj.sn. pr. inn- bygger ( %) |
| Oslo | 1989 | 950 | 130 |
| Østfold | 1714 | 409 | 112 |
| Hedmark | 1655 | 310 | 108 |
| Vest-Agder | 1616 | 240 | 106 |
| Oppland | 1595 | 292 | 105 |
| Buskerud | 1571 | 357 | 103 |
| Troms | 1541 | 231 | 101 |
| Nordland | 1536 | 370 | 101 |
| Telemark | 1530 | 250 | 100 |
| Nord-Trøndelag | 1516 | 194 | 99 |
| Sør-Trøndelag | 1491 | 381 | 98 |
| Aust-Agder | 1462 | 145 | 96 |
| Vestfold | 1438 | 290 | 94 |
| Hordaland | 1415 | 594 | 93 |
| Rogaland | 1365 | 479 | 89 |
| Sogn og Fjordane | 1358 | 146 | 89 |
| Møre og Romsdal | 1357 | 325 | 89 |
| Akershus | 1255 | 539 | 82 |
| Finnmark | 1245 | 95 | 82 |
| Sum/gjennomsnitt | 1526 | 6599 | 100 |
Kilde: NMD, 1995. NMD=Norsk Medisinaldepot; eneste grossist forut for 1.1.95.
Tabell 3.6 Utviklingen i aktørenes andeler av legemidler med utsalgspris fra apotek (AUP) på kr 150 og kr 500 i perioden 1.3.93 til 1.6.95
| Legemidler | Industri | Grossist (NMD) | Apotek | MVA | Sum |
| AUP = kr 150 pr. 1.3.93 | 55,9 | 5,9 | 20,2 | 18,0 | 100 |
| AUP = kr 150 +/- 20 % pr. 1.6.95 | 54,4 | 5,2 | 21,6 | 18,7 | 100 |
| AUP = kr 500 pr. 1.3.93 | 58,5 | 6,1 | 17,4 | 18,0 | 100 |
| AUP = kr 500 +/- 20 % pr. 1.6.95 | 61,5 | 4,6 | 15,2 | 18,7 | 100 |
Kilde: SHD 1994 og Konkurransetilsynet 1995.
Av tabell 3.6 kan vi se at produsentenes andel øker i forhold til de to øvrige aktører ved økende priser, spesielt i forhold til apotekvesenet. Dette har bl.a. sammenheng med den degressive prosentvise avansen i apotekvesenet.
Generelt viser utviklingen i perioden 1.3.93 til 1.6.95 at det ikke er store endringer i styrkeforholdet mellom aktørene. NMD har tapt andeler overfor produsenter for begge prisgrupper, og overfor apotek for den laveste prisgruppen. Dette synes rimelig på bakgrunn av NMDs avanseomlegging i løpet av 1994 hvor NMD fra 1. juni 1994 reduserte sin avanse i gjennomsnitt med 0,75 prosentenheter, og at det fra 1. desember 1994 ble foretatt en ytterligere omlegging av avansestrukturen samtidig som avansen i gjennomsnitt ble redusert med 3,5 prosentenheter (herunder avviklet NMD sin kontant- og ordrerabatt til apotekene på til sammen 2 prosentenheter). Videre fremgår det at apotekvesenet har økt sine andeler i pst. på billigere preparater, mens produsentene har økt sine andeler på dyrere preparater.
3.1.2 Internasjonalt
Norge har internasjonalt sett et lavt legemiddelforbruk, målt både i verdi- og volumtall. Ved vurdering av hva som er de mest brukte legemidlene i Norge kan en ta utgangspunkt i antall solgte doser eller pakninger av et legemiddel (volum), eller i omsetningsverdien målt i kroner.
Legemiddelforbruket målt i doser
Sammenligner en legemiddelforbruket knyttet til fire viktige legemiddelgrupper i doser i Norden for årene 1990-92, ligger Norge generelt lavere enn våre nordiske naboer Danmark og Sverige. Tabell 3.7 viser omsetningen av legemidler i Norge, Danmark og Sverige angitt i DDD pr. tusen innbyggere pr. døgn.
Tabell 3.7 Legemiddelforbruk i Norge, Danmark og Sverige (DDD/1 000 innbyggere/døgn)
| NORGE | DANMARK | SVERIGE | |
| Hjerte og kretsløp: | |||
| 1990 | 123,3 | 156,7 | 182,7 |
| 1991 | 126,6 | 161,7 | 181,4 |
| 1992 | 127,9 | 164,5 | 187,9 |
| Infeksjonssykdommer | |||
| 1990 | 14,4 | 11,2 | 17,3 |
| 1991 | 15,0 | 12,8 | 18,0 |
| 1992 | 15,6 | 13,9 | 19,8 |
| Muskel og skjelett | |||
| 1990 | 20,6 | 23,7 | 26,2 |
| 1991 | 20,8 | 26,4 | 27,8 |
| 1992 | 21,7 | 28,0 | 30,3 |
| Smertestillende | |||
| 1990 | 39,8 | 90,1 | 69,3 |
| 1991 | 40,1 | 91,2 | 68,8 |
| 1992 | 40,0 | 90,4 | 70,5 |
Kilde: «Nordic Statistics on Medicines 1990-1992»
Legemiddelforbruket målt i verdi
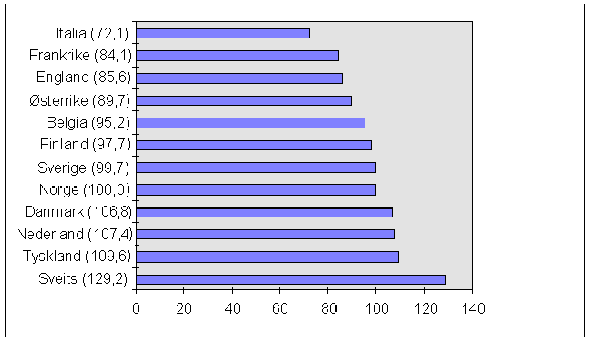
Japan har i dag verdens høyeste legemiddelutgift pr. innbygger, etterfulgt av USA og Frankrike. I et 20-års perspektiv har veksten i legemiddelutgiftene vært størst i Japan og USA. Beregnet ut fra grossistenes innkjøpspris (GIP) er legemiddelutgiftene pr. innbygger i Norge jf. figur 3.4 lavere enn i de fleste vesteuropeiske land. Alle våre nordiske naboland har høyere gjennomsnittlig legemiddelutgift pr. innbygger enn Norge. De eneste land i Vest-Europa som ligger lavere enn Norge, er Portugal, Hellas, Irland og England.
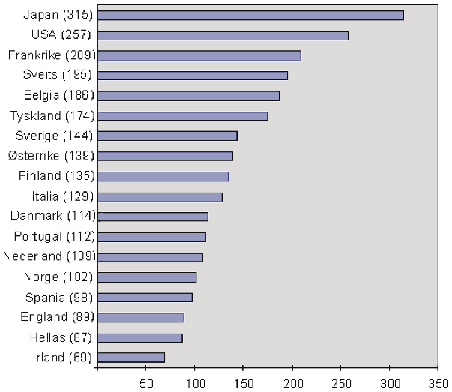
Figur 3.4 Legemiddelutgifter pr. innbygger, i Japan, USA og Vest-Europa 1993.
Indeks utregnet fra grossistenes innkjøpspris (GIP) og omregnet til ECU (kurs NOK 8,30)
Kilde: EFPIA, IMS og LMI, 1995.
Tallene påvirkes av pris- og kostnadsnivået i de enkelte land. Andre faktorer som kan bidra til variasjoner i legemiddelutgiftene er, terapitradisjoner, omfang og innretning av offentlige finansieringsordninger, kulturelle og demografiske trekk m.m.
Verdens legemiddelmarked utgjorde i 1995 236 mrd. US dollar regnet i grossistenes innkjøpspriser (GIP)(Kilde: IMS). De fattige land med 75 pst. av verdens befolkning i 1995 forbruker under 20 pst. av verdens legemidler. De 20 største legemiddelmarkedene i verden står for over 80 pst. av legemiddelomsetningen. USA hadde i 1994 et forbruk av legemidler som tilsvarer 32 pst. av legemiddelmarkedet. Japan har en markedsandel på 21 pst. og hele Vest-Europa 28 pst.
(Kilde: IMS).
Prisnivå på legemidler
Figur 3.5 Prisnivået på legemidler i noen europeiske land, 1995 (Norge=100)*)
*) Prisindeksen er beregnet ut fra GIP-priser og svensk volum i tidsrommet 1.1.95-31.12.95, og valutakurser pr. 2.1.96. GIP-prisene er beregnet ut fra de respektive lands apotekprislister, justert med gjennomsnittlige grossistmarginer.
Kilde: Apoteksbolaget, Sverige (Tallene er omregnet til Norge=100)
I en undersøkelse utført av det svenske Apoteksbolaget, er det foretatt sammenligninger av prisen (GIP) på de 408 mest solgte legemidlene i Sverige med prisene i 11 andre europeiske land. 290 av de 408 legemidlene på det svenske markedet fantes også på det norske. Tradisjonelle lavprismarkeder som Hellas, Portugal og Spania er ikke tatt med i undersøkelsen. Det fremgår at prisene på legemidler i Norge er høyere enn i Finland, men lavere enn i Danmark.
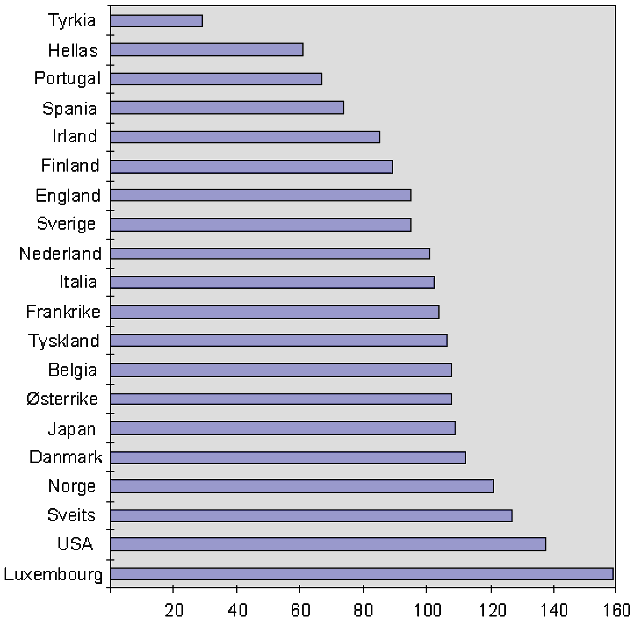
Figur 3.6 Kjøpekraftsnivå internasjonalt, 1995
Kjøpekraftskorrigert indeks for BNP pr. innbygger, OECD = 100
Kilde: OECD, Main Economic Indicators, juni 1996
Figuren gir et bilde av den ulike kjøpekraftsituasjonen i EØS-området. Det fremgår ved å sammenlikne figurene 3.5 og 3.6 at kjøpekraften gjennomgående er høy i høyprisland, men det er også klare unntak fra denne regelen, som f.eks. Frankrike (høy kjøpekraft, men med lave priser) og Nederland (under middels, men med høye priser). Spania, Portugal og Hellas har et lavt kjøpekraftsnivå. Disse land har også lave priser, og er i stor grad utgangspunkt for parallelleksport mot land i Nord-Europa.
3.2 GJELDENDE REGULERINGER INNEN LEGEMIDDELSEKTOREN
3.2.1 Målsettinger for myndighetenes reguleringspolitikk på legemiddelområdet
Den norske legemiddelpolitikken er formet over mange tiår og reflekteres i lover, forskrifter og i viktige helsepolitiske veivalg.
I Stortingsmelding nr. 41 (1987-88), Helsepolitikken mot år 2000, gjentatt i Stortingsproposisjon nr. 1 (1995-96), Sosial- og helsedepartementet, med unntak av ens legemiddelpriser som ikke lenger gjelder (maksimalprisregulering), ble følgende målsettinger lagt frem:
befolkningen i alle deler av landet skal få rimelig lett og sikker adgang til effektive og sikre legemidler til lavest mulig pris og med ens utsalgspris over hele landet
ressursene skal være hensiktsmessig fordelt innenfor legemiddelsektoren
det skal sikres en forsvarlig og medisinsk riktig (rasjonell) legemiddelbruk, så vel i som utenfor institusjon
3.2.2 Offentlige reguleringstiltak i markedet for legemidler
Sentrale myndigheter har i utstrakt grad benyttet reguleringstiltak i legemiddelmarkedet med sikte på å realisere overordnede målsettinger for legemiddelpolitikken. Sentrale virkemidler i denne sammenheng har vært godkjenning av legemidler og utstedelse av markedsføringstillatelse for det norske marked, fastsettelse av apotekavanse og legemiddelpriser (fra 1.1.95 bare for reseptpliktige legemidler), krav til utøvelse av distribusjonsvirksomheten, behovsprøving for apoteketablering, trygderefusjon for legemidler på blå resept, krav til sortimentsbredde og landsdekning for grossister og leveringsplikt i apotek. Myndighetenes reguleringstiltak følger videre nærmere omtalt.
3.2.2.1 Godkjenning av legemidler i Norge
Alle legemidler eller farmasøytiske spesialpreparater (dvs. legemidler som bringes i handelen i produsentens originale pakning og med eget preparatnavn) skal være godkjent og ha markedsføringstillatelse. Markedsføringstillatelse erstatter det som tidligere ble kalt «registrering», og omtales nærmere nedenfor.
Selve godkjenningsprosedyren foregår i flere trinn. Legemiddelfirmaet sender søknad med dokumentasjon (kvalitet, sikkerhet og effekt) til Statens legemiddelkontroll sammen med betalt avgift. Denne avgiften er ment å skulle dekke utgiftene ved behandlingen av søknader. Dokumentasjonens tre deler blir utredet hver for seg. Søkeren får tilsendt evalueringsprotokollen og har anledning til å gi kommentar. I vurderingen av søknaden vil kravene til dokumentert sikkerhet, kvalitet og effekt avgjøre utfallet. Sikkerhet, kvalitet og effekt var også kriteriene for «medisinsk berettigelse», som inntil opphevelsen av behovsparagrafen (omtalt under 3.2.2.2), var et krav for godkjenning av legemidler. Omleggingen i forbindelse med opphevelse av behovsparagrafen innebar således ikke at kravene til dokumentasjon ble endret.
Søknader om nye virkestoffer eller utvidelser av bruksområder for godkjente legemidler, går videre til Spesialitetsnemnda for råd, før vedtaket fattes av Statens legemiddelkontroll. Som ledd i omorganiseringen innen helseforvaltningen, er mandatet for Spesialitetsnemnda endret. Nemnda har ikke lenger besluttende myndighet, men skal gi råd til Statens legemiddelkontroll i spørsmål om godkjenning av nye virkestoff, nye indikasjoner og tilbaketrekking av markedsføringstillatelser. Statens legemiddelkontroll trekker således på den faglige kompetanse i Spesialitetsnemnda.
Dersom vurderingen er negativ på ett eller flere av de søkte bruksområder, har søkeren 6 måneder til å supplere søknaden. Søkeren kan også velge å trekke søknaden.
Godkjenning av parallellimporterte legemidler
Parallellimport av legemidler til Norge innebærer at et preparat som allerede har norsk markedsføringstillatelse (som regel et patentert produkt) importeres fra et land med lavere prisnivå. Den som importerer preparatet, parallellimportøren, selger da preparatet på det norske markedet i konkurranse med orginalprodusentens direkteimporterte preparat og dennes salgsrepresentant i landet. Alle parallellimporterte legemidler må godkjennes av Statens legemiddelkontroll etter forenklet prosedyre, og ingen legemidler kan selges i Norge uten markedsføringstillatelse. Legemiddelkontrollens viktigste oppgave, før markedsføringstillatelse utstedes, er å kontrollere at det ikke er terapeutiske forskjeller av betydning mellom parallell- og direkteimporterte legemidler.
Tillatelse utstedes etter en vurdering av preparatet ved Statens legemiddelkontroll. Når det gjelder krav til effekt, forutsettes det dokumentert ved vurdering av det direkteimporterte legemidlet. Etiketten/merkingen av legemidlene skal også være i tråd med norske krav. Det vil si at alle opplysninger skal gis på norsk. I tillegg til dette kan utenlandsk tekst aksepteres, men aldri alene.
Godkjenning av vaksiner og andre biologiske legemidler
Tidligere fantes det ikke offentlige kvalitetsstandarder for vaksiner i Norden. Krav til kvalitet ble i Norge forvaltet av Statens institutt for folkehelse og Veterinærinstituttet. Disse var også produsenter og kontrollører av utenlandske preparater. Nå blir vaksiner godkjent som farmasøytiske spesialpreparater av Statens legemiddelkontroll.
For legemidler av biologisk opprinnelse er det spesielt viktig med standardkrav til prosesskontroll, fordi styring av selve fremstillingsprosessen er helt avgjørende for kvaliteten av det ferdige produktet. Dette gjelder for vaksiner og f.eks. insulin, veksthormon og blodfaktorer.
Samarbeidet i Norden og i Den europeiske farmakopékommisjon har vært relatert til standardutvikling. Den europeiske farmakopékommisjonen ligger under Public Health Committee, som igjen er et organ under Europarådet. Farmakopékommisjonen godkjenner legemiddelstandarder etter råd fra sine ekspertgrupper. Implementeringen skjer gjennom Public Health Committee. Den europeiske farmakopé (Ph. Eur.) er en samling autoriserte standarder for legemidler (ferdige legemidler, råstoffer og analysemetoder).
Godkjenning av legemidler fremstilt ved moderne bioteknologi
Det biologiske grunnlaget for bioteknologien, samt bioteknologiske teknikker og prosesser, er kortfattet beskrevet i Stortingsmelding nr. 8 (1990-91), Om bioteknologi.
Bioteknologi brukes til å fremstille legemidler av biologisk opprinnelse i industriell skala, i likhet med kjemiske metoder som brukes for å fremstille farmasøytisk-kjemiske legemidler. Selve behandlingen blir som ved et hvilket som helst annet legemiddel.
Ved genteknikk oppnår man et veldefinert legemiddel idet den genetiske koden sikrer at det produseres eksakte kopier av det naturlig forekommende protein, der dette er tilsiktet, og man oppnår en høyere renhetsgrad enn ved konvensjonell produksjon. Dette innebærer også at den rutinemessige kontroll av preparatets biologiske aktivitet ikke behøver å baseres på biologisk styrkebestemmelse, men kan utføres med fysikalsk-kjemiske eller immunologiske metoder. En annen fordel er at man unngår risiko for eventuell smitte fra mulig forurenset biologisk utgangsmateriale (f.eks. humant blod).
Mange av de nye legemidlene som etterhvert fremstilles med genteknikk, har tidligere ikke vært tilgjengelige fordi de forekommer i så lave konsentrasjoner i biologisk materiale at det ikke har vært mulig med fremstilling i stor skala. Denne begrensningen er overvunnet ved hjelp av genteknologien.
Legemiddelsamarbeidet i EU har resultert i retningslinjer for produksjon og kontroll av legemidler fremstilt ved moderne bioteknologi. I henhold til EØS-avtalen gjelder disse retningslinjene i Norge. Retningslinjene ligger også til grunn for utarbeidelsen av det dokumentasjonsmateriale for produktkvalitet som produsenten skal sende inn i forbindelse med søknad om markedsføringstillatelse.
Godkjenning av naturlegemidler
Det finnes ikke noe eget EU-direktiv som omhandler «naturmidler». Dersom man ønsker å presentere et «naturmiddel» som et middel mot sykdom, eller tar sikte på bruk av dette som et diagnostisk middel eller til å påvirke legemsfunksjoner, faller det innenfor definisjonen av et legemiddel. Midlet må dermed dokumenteres og søkes godkjent som et ordinært legemiddel eller et naturlegemiddel. Det kan ikke legalt markedsføres med påstander om forebyggende, lindrende eller legende effekt på sykdom, smerte eller den fysiologiske prosessen i kroppen før markedsføringstillatelse som legemiddel eller naturlegemiddel er oppnådd. I markedsføringstillatelsen vil det være definert klare rammer for hvilke påstander om preparatet som kan benyttes i markedsføringen.
Godkjenningsordningen i Norge baserer seg på at produkter som konsumeres av mennesker enten er næringsmidler (ev. kosmetikk hvis de benyttes utvortes) eller legemidler. Naturmidler kan godkjennes som farmasøytiske spesialpreparater, såkalte naturlegemidler, etter en forenklet prosedyre. Det er utarbeidet retningslinjer for godkjenning av naturlegemidler. Kravene til kvalitet av naturlegemidler er like omfattende som for ethvert annet farmasøytisk spesialpreparat. Det er ingen krav til vitenskapelig dokumentasjon av sikkerhet og effekt, men dokumentasjonen skal redegjøre for erfaringer fra tradisjonell bruk i folkemedisin i Europa eller Nord-Amerika som viser at preparatet er ufarlig i bruk.
3.2.2.2 Markedsføringstillatelse
Når Statens legemiddelkontroll har godkjent et legemiddel ut fra vurdering av dets sikkerhet, kvalitet og effekt, må det utstedes markedsføringstillatelse før legemidlet tillates solgt. Dette betyr i praksis at preparatets navn, pakningsstørrelse, teknisk utstyr, emballasje, merking, pakningsvedlegg og preparatomtale skal godkjennes. Videre skal det fattes avgjørelser vedrørende reseptplikt og reseptgruppe, og om eventuelle særlige utleveringsbestemmelser eller forskrivningsregler. Når markedsføringstillatelse er utstedt, føres legemidlet opp i Spesialitetsregisteret som omfatter de til enhver tid godkjente legemidler med markedsføringstillatelse. Innehaver av markedsføringstillatelse er forpliktet overfor gjeldende bestemmelser blant annet om innførsel, markedsføring, reklame og klinisk utprøving. Markedsføringstillatelse omfatter ikke prisgodkjenning.
Godkjenningsfritak for legemidler
Lov om legemidler mv. av 4. desember 1992 gir adgang til at leger, tannleger og veterinærer i en viss utstrekning kan skrive ut resepter på legemidler uten markedsføringstillatelse i Norge. Denne ordningen skal sikre et behandlingstilbud til pasienter som ikke får tilfredsstillende tilbud gjennom de legemidler som er markedsført i Norge. Det er mulig å forskrive til enkeltpasienter eller til sykehusavdelinger.
Søknader om utlevering av preparater uten markedsføringstillatelse vurderes av Statens legemiddelkontroll. Det foreligger ofte sparsom dokumentasjon for slike preparater hos Statens legemiddelkontroll, og det er derfor forskrivers ansvar å forsikre seg om at kvalitet, sikkerhet og effekt er tilstrekkelig. For enkelte typer preparater settes særlige utleveringsrestriksjoner av sikkerhetsmessige grunner.
Det kommer årlig inn mellom 20 000 og 25 000 slike søknader, det vil si en av tusen resepter. I praksis blir slike søknader sjelden avslått.
EUs godkjenningssystem
EU har etablert et nytt system for godkjenning av legemidler. Nye bioteknologiske legemidler skal alltid godkjennes i en sentral prosedyre. Andre preparater (bl.a. preparater med høy innovasjonsverdi) kan også godkjennes i den sentrale prosedyren etter vurdering av EU (liste b-preparater). Nye legemidler kan også godkjennes i alle EU-land på basis av godkjenning gitt i ett land. Det nye systemet introduserer nye prosedyrer og institusjoner i godkjenningsarbeidet. Systemet endrer ikke kriteriene for godkjenning av legemidler. Kravene om dokumentert effekt, sikkerhet og kvalitet ligger fast. Norge har ikke del i ordningen, som trådte i kraft ved årsskiftet 1994-95.
Det europeiske legemiddelkontoret EMEA (European Medicines Evaluation Agency) er den sentrale administrative enheten i systemet. Det legemiddelfaglige arbeidet gjøres i to fagkomiteer som hver har to representanter fra hvert av EU-landenes kontrollmyndigheter: CPMP (Committee for Proprietary Medicinal Products) tar seg av humane legemidler, mens CVPM (Committee for Veterinary Medicinal Products) har hånd om legemidler til dyr.
Som søkerland til EU deltok Norge med observatører i fagkomiteer og arbeidsgrupper som inngår i det nye systemet. Norge deltar siden sommeren 1996 gjennom Nordisk legemiddelnemnd som observatører i flere arbeidsgrupper, men vi har ikke adgang til det legemiddelfaglige arbeidet i EU når det gjelder enkeltpreparater. Norge deltar fremdeles i arbeidet i den rådgivende delen i EU via Pharmaceutical Committee. Dette er hjemlet i EØS-avtalen. Den farmasøytiske komité er rådgivende for kommisjonen i lovspørsmål og skal behandle anvendelse av direktivene på legemiddelområdet og forslag til nye direktiver.
Virkningene av EØS-avtalen for det norske godkjenningssystemet
Foruten legemiddelloven er en rekke forskrifter, retningslinjer og veiledninger revidert som følge av EØS-avtalen. I arbeidet med godkjenning av legemidler er bortfallet av den såkalte behovsparagrafen den mest betydningsfulle av endringene. Andre endringer er mest av terminologisk og praktisk art.
Opphevelsen av behovsparagrafen
I 1938 ble den såkalte «behovsparagrafen» innført. Behovsparagrafen ga norske legemiddelmyndigheter anledning til å holde utvalget av godkjente legemidler på et oversiktlig nivå, ved å avvise legemidler som det ikke var et medisinsk behov for.
Blant annet har ordningen vært brukt til å begrense utvalget av legemidler. Norge har også hatt krav om fornying av godkjenning for farmasøytiske spesialpreparater hvert 5. år. Dette har gjort det mulig å avregistrere legemidler uten dokumentert effekt. En slik bestemmelse er nå innført i EU-landene.
Behovsparagrafen ble også anvendt overfor legemidler som innfridde de faglige krav knyttet til effekt, sikkerhet og kvalitet. Denne praktisering av behovsparagrafen ble derfor vurdert som et teknisk handelshinder og ordningen falt bort som følge av EØS-avtalen.
Søknader om markedsføringstillatelse med og uten behovsparagraf
Desember 1992 ble ny lov om legemidler uten behovsparagraf vedtatt. I hele 1993 var det usikkerhet om når EØS-avtalen, og derved den nye legemiddelloven, skulle tre i kraft. Det skjedde 1.1.94. Usikkerheten i 1993 om når behovsparagrafen ville bli opphevet, gjør at 1992 er valgt som sammenligningsgrunnlag for å vise konsekvensene av at behovsparagrafen er opphevet.
Krav om markedsføringstillatelse for radiofarmaka, veterinærvaksiner og naturlegemidler er innført i denne perioden, og parallellimport er blitt aktuelt. Søknader for slike preparater (til sammen 188 i 1994) er ikke tatt med i tabell 3.8.
Tabell 3.8 Utviklingen i søknader om registrering/godkjenning i perioden 1992-94
| Nyhetsverdi | ||||
| liten | middels | Stor | I alt | |
| Antall nye virkestoffer i 1992 | 40 | 10 | 0 | 50 |
| Antall nye virkestoffer i 1994 | 50 | 6 | 1 | 57 |
Kilde: SLK, 1995-96.
Antall søknader har økt etter at behovsparagrafen ble opphevet. Økningen skyldes først og fremst flere søknader for synonympreparater, men det er også en viss økning i nye virkestoffer, som vurderes av SLK til å ha liten nyhetsverdi i terapeutisk sammenheng.
3.2.2.3 Prisfastsettelse på legemidler
Historisk oversikt
Allerede forordningen av 1619 med generelle bestemmelser om apotekdrift fastslo at det skulle utarbeides en offentlig takstfortegnelse til bruk i apotek. Taksten ble fornyet når forholdene gjorde det påkrevet, men selve råvareprisene kunne korrigeres i takt med anskaffelsesprisen etter individuelle avtaler mellom apoteker og berørte leger.
Prinsippet om ens legemiddelpriser over hele landet ble innført fra 1806. To år senere ble det påbudt at prisen, samt apotekets og ekspeditørens navn, skulle settes på alle ekspederte resepter. Det ble også fastsatt straff for overtredelse av takstene. Prisforskriften og en offentlig takstbok, medisinaltaksten, utarbeidet på grunnlag av reglene i prisforskriften, har således regulert apotekenes prissetting gjennom mange år.
Fra 1891 måtte apotekene sende deler av driftsregnskapet til departementet. Dette gav myndighetene innsyn i bransjens økonomiske forhold og samtidig grunnlaget for regulering av legemiddelprisene.
Mot slutten av 1800-tallet skapte utviklingen av industrifremstilte legemidler problemer for forsvarlig prissetting. Fra 1904 kunne Kongen forby omsetning av legemidler hvis prisen stod sterkt i misforhold til verdien av innholdsstoffene. I 1928 ble det etablert offentlig prisgodkjenning av farmasøytiske spesialpreparater.
Dagens praksis
Fra 1.1.95 ble ny forskrift om prisfastsettelse på legemidler tatt i bruk (Forskrift om prisfastsettelse av legemidler). Denne forskriften regulerer alle reseptpliktige legemidler som omsettes i Norge. De reseptfrie legemidlene blir ikke lenger prisregulert.
Før et reseptbelagt legemiddel bringes i handelen eller omsettes, skal dets maksimale utsalgspris fra apotek (AUP) fastsettes av Sosial- og helsedepartementet eller den det bemyndiger.
Fastsettelse av maksimal AIP-pris for reseptbelagte farmasøytiske spesialpreparater med registrert markedsføringstillatelse (MT nr.) er delegert til Statens legemiddelkontroll (SLK). Statens helsetilsyn fastsetter maksimal avanse for apotek ved omsetning av reseptbelagte legemidler. På grunnlag av maksimal AIP og maksimal apotekavanse fastsetter SLK maksimal apotekutsalgspris (AUP).
Å gi bestemmelser om maksimalpriser for reseptbelagte legemidler som produseres i apotek er delegert til Statens helsetilsyn.
SLK skal informere apotekene om gjeldende maksimalpriser.
Prosedyrer for prisfastsettelse av reseptpliktige legemidler
Et reseptpliktig farmasøytisk spesialpreparat som har fått markedsføringstillatelse i Norge (MT nr.) kan ikke omsettes før det har fått fastsatt maksimal utsalgspris fra apotek (AUP). MT nr. gis for det enkelte preparat bestemt ved merkenavn, legemiddelform og styrke. Maksimalpris fastsettes for den enkelte godkjente pakningsstørrelse.
Prisforskriften gir denne bestemmelse om prisfastsettingen (§ 2):
«Ved fastsetting av prisen for et farmasøytisk spesialpreparat skal det påses at den ikke står i misforhold til verdien. Herunder tas hensyn til prisen for preparat med tilsvarende effekt eller lignende virkestoff, prisen på preparatet i andre EU/EØS-land og til opplysninger om produksjonskostnadene for preparatet.»
Produsenten skal gi forslag til AIP-pris, som vurderes av SLK. Produsenten skal dessuten gi opplysning om priser i andre EØS-land. Produsenten gir selv andre opplysninger som kan underbygge prisforslaget.
SLK vurderer produsentens prisforslag i samsvar med prisforskriften:
preparatets verdi Preparatets verdi som legemiddel er den overordnede vurdering. Denne vurdering bygger på konklusjonene i de evalueringsrapporter som foreligger for godkjennelse av preparatet
andre preparater med tilsvarende effekt Det vil gå frem av nevnte evalueringsrapporter om preparatet har lik, tilsvarende eller vesentlig annerledes effekt enn preparater på markedet som er godkjent for den samme lidelse
andre preparater med lignende virkestoff Om preparat med lignende virkestoff er på markedet, i tilfelle hva avviket består i, vil gå frem av de nevnte rapporter
preparatets priser i andre EØS-land
Produsenten gir opplysninger om dette
opplysninger om produksjonskostnadene Det utbes opplysninger om produksjonskostnader i konkrete saker der dette kan få betydning for prisvurderingen
Det vil nesten alltid foreligge en grad av likhet til et alternativt preparat på markedet eller annen behandling for lidelsen.
Når produsenten foreslår en høyere pris enn på de nærmest tilsvarende preparater, begrunnes dette med at preparatet er bedre, eller med at prisene ligger på dette nivå i andre land. SLK vurderer forskjellene på grunnlag av de nevnte evalueringsrapporter. Det er vanligst at preparatene er kjemisk eller teknisk forskjellige. Det avgjørende for prisvurderingen er den verdi slike forskjeller har for preparatet som legemiddel.
Ved begrunnet krav om et høyere prisnivå skal forslaget fra produsentene sammenlignes med priser i andre EØS-land. Disse opplysninger kan gi argumenter for å holde prisnivået, eller akseptere en høyere pris.
Noen nye preparater innebærer en så vesentlig bedre effekt for pasienten at de preparater som hittil er brukt ved lidelsen egentlig ikke er et alternativ når den nye behandlingen foreligger. I slike tilfeller vil det bl.a. legges vekt på hvilken pris produsenten har oppnådd i andre land for legemidlet.
Behandlingstiden for prissaker skal være høyst 90 dager, ifølge prisforskriften.
Prisfastsettelser som ligger lavere enn produsentens forslag kan påklages i henhold til forvaltningsloven. SLK kan etter ny vurdering endre vedtaket, eller saken kan ekspederes til Sosial- og helsedepartementet, som er klageinstans.
Prisjustering på reseptpliktige legemidler som er på markedet
Prisforskriften gir SLK hjemmel til å styre søknader om prisjusteringer på generelt grunnlag til en gang pr. år, kalt «årlige prisjusteringer». Dette gir mulighet for en enhetlig vurdering av grunnlaget for slike søknader. Slike eventuelle prisjusteringer skal foretas på grunnlag av objektive kostnadskriterier, f.eks. valutakursendringer, kostnadsforskyvninger og prisendringer. Forut for den søknadsfrist som fastsettes, vil kriteriene for søknad bli drøftet med produsentene.
Prisforskriftens § 4 hjemler partenes adgang til å ta opp spørsmålet om prisendringer dersom endrede forhold eller nye opplysninger tilsier det. Denne bestemmelsen gjør det bl.a. mulig å fastsette lovlig høyere priser for preparater som produsenten ellers ville trekke fra markedet på grunn av for lav pris. Bestemmelsen gir også hjemmel for å kreve prisreduksjon. Ingen legemidler har vært satt ned i pris med hjemmel i denne bestemmelsen.
I perioden 1980 til 1993 ble «årlige prisjusteringer» avtalt i forhandlinger mellom Sosialdepartementet og industriens organisasjoner. Enkelte år ble den praktiske gjennomføring av forhandlingene delegert til daværende Helsedirektoratet. I forhandlingene ble det fastlagt hvilke kostnadsforhold og hvilke betingelser som skulle legges til grunn for prisjusteringer. Som dokumentasjon for kostnadsforholdene ble det benyttet offisiell statistikk for utviklingen i de enkelte produsentland og i Norge. For 1993 og 1994 forhandlet Helsedirektoratet både med innen- og utenlandsk industri. Som resultat ble det fastsatt et likt tillegg, ett for norske og ett for utenlandske produsenter.
Gevinstdelingsmodell for apotek
Dersom faktisk AIP (etter at rabatter er trukket fra) er lavere enn fastsatt maksimal AIP, skal fastsatte avansesatser benyttes på faktisk innkjøpspris.
Ved gevinstdeling fremkommer AUP ved først å legge til ordinær avanse på faktisk AIP og deretter legge til maksimalt halvparten av differansen mellom maksimal AUP og faktisk AUP beregnet etter ovennevnte avanser (eks. mva.). Til sist tillegges mva. Minst halvparten av differansen mellom maksimal AIP og faktisk AIP skal tilfalle kunden.
Kontroll med prisene i apotek
Det har generelt ikke vært kontroll med prisene i apotek fra Helsetilsynets side. Legemiddelinspektørene har i den senere tid gjort stikkprøvekontroller for å se hvordan den nye prisforskriften med krav til rabattdeling blir praktisert.
Endring i pris/refusjonsregler som følge av EØS
Bestemmelsene om prising av legemidler til mennesker og fastsettelse av refusjonsregler for legemidler på «blå resept» er å finne i Direktiv 89/105/EF, det såkalte «transparency-direktivet».
Transparency-direktivet om innsyn i prisbestemmelser for legemidler og slike legemidlers opptak i de nasjonale syketrygdordninger skal sikre at nasjonale prisordninger, herunder hvordan de virker i enkeltsaker og de kriterier de bygger på, er offentlig tilgjengelige for alle deltagere i legemiddelmarkedet. Som et første skritt for å fjerne ulikhetene landene imellom er det fastlagt en rekke krav som gjør det mulig for alle berørte parter å kontrollere at nasjonale tiltak ikke innebærer kvantitative restriksjoner på import eller eksport, eller tiltak med tilsvarende virkning. EU sikter mot like konkurransevilkår også på legemiddelområdet gjennom å etablere et felles marked for legemidler. Kravene i direktivet får ingen innvirkning på medlemsstatenes politikk når det gjelder prisfastsettelse og innføring av trygdeordninger, med mindre dette er nødvendig for å oppnå innsyn.
3.2.2.4 Reklamebestemmelser
Dagens norske regulering av reklame for legemidler
De norske forskriftene trådte i kraft 25. august 1994, og er basert på EUs Rådsdirektiv 92/28/EØF og samsvarer i store trekk med de gamle forskriftene. Grunnregelen for all legemiddelreklame er at den skal være nøktern og saklig. Forskriften skiller mellom reklame til allmennheten og reklame til helsepersonell. Til allmennheten kan det kun reklameres for reseptfrie legemidler. Slik reklame skal ikke virke støtende og den skal bl.a. inneholde forsiktighetsregler. Reklame for reseptbelagte legemidler kan kun rettes mot helsepersonell og skal inneholde elementer som i stor grad sammenfaller med den godkjente preparatomtale. Forskriften inneholder også en rekke andre bestemmelser.
Forskriftene innebærer også at tidligere forbud mot reklame for reseptfrie legemidler i radio og på offentlig sted (trafikkreklame, kino-/teaterreklame m.m.) og forbud mot reklametrykksaker (direkte reklame) er opphevet, mens reklame for legemidler fortsatt er forbudt på fjernsyn.
Statens legemiddelkontroll skal fortsatt overvåke reklame for legemidler, men det er ikke lenger krav om forhåndsgodkjenning. Ved overtredelse av bestemmelsene kan det kreves at reklamen stoppes, og at det sendes ut en beriktigelse. Ved gjentatte overtredelser kan Statens legemiddelkontroll forby all reklame for vedkommende produkt for kortere eller lengre tid. Vedtakene offentliggjøres.
Avtale med Legemiddelindustriforeningen om tilsyn med reklame for legemidler
Forhåndsgodkjenning av reklame gjøres ikke lenger av myndighetene. Firmaene må selv ta det fulle ansvar for at markedsføringen er i samsvar med forskriftene og sørge for at de benytter den nødvendige ekspertise til å foreta denne vurderingen før de går ut med markedsføring.
Pr. 1. oktober 1994 inngikk Statens legemiddelkontroll en avtale med Legemiddelindustriforeningen om at Rådet for vurdering av legemiddelinformasjon («Rådet») løpende vurderer om legemiddelreklamen er i samsvar med den nye reklameforskriften. Rådet arbeider uavhengig av Legemiddelindustriforeningen.
3.2.2.5 Patentregler
Patentlovgivningen ivaretar hensynet til innovativ legemiddelindustri ved at originalpreparater beskyttes mot konkurranse fra kopiprodukter. Gjennom denne beskyttelsen, og gjennom den etterspørsel et originalprodukt er i stand til å generere i markedet på grunnlag av preparatets pris og kvalitet, har forskningsbaserte legemiddelselskaper mulighet til å tjene inn sine forsknings- og utviklingskostnader. Det vises til kapittel 6 for nærmere drøfting av patentordningen.
3.2.2.6 Reguleringer av grossist-/importvirksomhet
Regulering av grossist-/importvirksomhet i henhold til EØS-avtalen
Innen EU er reglene for grossistvirksomhet med legemidler fastsatt i direktiv 92/25. Virksomheten omfattes også av andre deler av EUs regelverk, bl.a. konkurransereglene. Det nevnte grossistdirektiv er inkludert i tilleggsavtalen til EØS-avtalen.
Grossistvirksomhet med legemidler defineres som alle aktiviteter som innebærer anskaffelse (herunder import), lagring, forsyning og eksport av farmasøytiske produkter, med eller mellom produsenter, agenter, grossister eller apotek.
Grossistvirksomheten krever tillatelse gitt av nasjonal myndighet. Det enkelte land må respektere tillatelser gitt av andre lands myndigheter (EØS-området). Myndighetene i det enkelte land kan pålegge grossistene en spesiell offentlig tjenesteplikt. Det er imidlertid en forutsetning at denne tjenesteplikten ikke blir utnyttet til fordel for nasjonale grossister.
Med EØS-avtalen ble det (fra 1. januar 1995) åpnet opp for at firmaer med tilvirkertillatelse innen EØS-området kan opptre som grossister for egne produkter. Videre ble NMDs enerett til import (eksport) og engrosomsetning opphevet. Parallellimport (eksport) av legemidler ble tillatt i Norge fra 1. januar 1994 og for legemidler med gyldig patent fra 1. januar 1995
Regulering av det norske markedet
Myndighetene i det enkelte land har etter EØS-reglene mulighet til å pålegge grossistene «å garantere et tilstrekkelig utvalg av legemidler som til enhver tid kan dekke behovene i et bestemt geografisk område, og til å sikre levering av de forsyninger det anmodes om i løpet av svært kort tid over hele det aktuelle området». Norske myndigheter har benyttet seg av denne retten for å sikre at alle brukere i Norge skal ha samme tilgang til legemidler i hele landet.
I desember 1993 forelå den norske forskriften (Forskrift om grossistvirksomhet med legemidler) på området. Forskriften er konkretisert i retningslinjer for utøvelse av import- og grossistvirksomhet med legemidler.
Den norske forskriften omfatter grossistvirksomhet med både human- og veterinærpreparater. EØS-direktivet gjelder kun humanpreparater.
Krav til leveringsdyktighet:
grossister må føre det utvalg av legemidler som etterspørres i det norske markedet (§ 4) (krav til sortimentsbredde)
grossister må som hovedregel kunne levere hvor som helst i landet innen 24 timer, for spesielt vanskelig tilgjengelige områder er fristen 48 timer (§ 4)
Disse kravene kan det gjøres unntak for når særlige grunner foreligger.
Andre sentrale bestemmelser i forskriften er:
firmaer med tilvirkertillatelse innen EØS-området kan fra 1. januar 1995 opptre som grossister for egne produkter (§ 3)
kvalifisert person (cand. pharm.) skal være ansvarlig for den farmasøytisk-faglige delen av virksomheten
Tilvirkere har automatisk rett til å opptre som grossister for de preparater som omfattes av tilvirkertillatelsen. Kravet om levering av alle preparater som etterspørres i det norske marked gjelder ikke for disse. Det er samme krav til landsdekkende levering innen 24 (48) timer, som for andre grossister.
Fra og med 1. november 1995 har Norge tre grossister som tilfredsstiller alle krav i grossistforskriften.
Parallellimport av legemidler
Parallellimport er hjemlet i EUs prinsipp om fri flyt av varer og er i dag mulig i hele EU- og EØS-området. I Norge er forholdet regulert i Lov om legemidler, forskriften om grossistvirksomhet og retningslinjer for ervervsmessig inn- og utførsel av legemidler. Preparatet som det parallellimporterte preparatet søkes importert i konkurranse med, kalles direkteimportert preparat. Begrepet direkteimportert anvendes uavhengig av om preparatet er importert eller tilvirket i Norge. Dersom det direkteimporterte preparatet trekkes fra det norske markedet, faller også grunnlaget for parallellimport bort.
Forskriften om grossistvirksomhet tillater i §1 import foretatt av importør som er godkjent av Statens helsetilsyn. Retningslinjer for ervervsmessig inn- og utførsel av legemidler gir innehaveren av importørgodkjenning rett til å:
importere legemidler, inkludert parallellpreparater til Norge
lagre legemidlene
selge legemidlene til godkjent grossist
selge aktive substanser til tilvirkere
eksportere legemidlene fra Norge
3.2.2.7 Direktesalg til sykehus og andre helseinstitusjoner
I samsvar med tidligere lovverk skulle legemidler som hovedregel omsettes gjennom grossist og apotek. Norsk Medisinaldepot har hatt tillatelse til å selge følgende preparatgrupper direkte til sykehus og sykehjem:
dialysevæsker
hemodialysekonsentrater
infusjonsvæsker
skyllevæsker
sondeernæringspreparater i bruksferdig form
Bakgrunnen for denne tillatelsen var at dette ble sett på som særlig plasskrevende legemidler, som det var praktisk å levere direkte til sykehusene. Forutsetningen var at helseinstitusjonene hadde en størrelse som tilsvarte minimum 50 senger.
I forbindelse med EØS-avtalen har Norge forpliktet seg til å følge EUs regler angående offentlige innkjøp. Regelverket gjelder også for legemidler. I tråd med forslag til Ot.prp. nr. 3 (1994-95) Endringer i lov av 17. juni 1966 nr. 12 om folketrygd og i visse andre lover, ble det i 1995 åpnet for direktekjøp av legemidler fra grossist/produsent til sykehus for utvalgte legemiddelgrupper som sykehusene er storbrukere av. Formålet var å oppnå reduserte legemiddelkostnader, og å gjøre det enklere for sykehusene å praktisere anbud ved innkjøp. Omleggingen, som innebar en opphevelse av apotekenes enerett for sykehussalg, var ikke en konsekvens av EØS-avtalen. Ved omleggingen ble det slått fast at den faglige standard knyttet til håndtering av legemidler ikke skulle svekkes.
Sykehus som har etablert en farmasøytisk avdeling i tråd med forskriften om legemiddelforsyningen mv. ved sykehus og andre helseinstitusjoner, og som kan dokumentere at den farmasøytiske kvalitet opprettholdes i forhold til eksisterende leveringsordning, kan kjøpe inn direkte fra grossist/produsent de legemiddelgrupper som fremgår av en spesifisert liste. Sykehus som ikke har farmasøytisk avdeling kan kjøpe inn legemidler etter den ordning som hittil har vært gjeldende.
Som følge av EØS-avtalens bestemmelser om anbudsinnkjøp til offentlige institusjoner, har 17 fylkeskommuner gått sammen om et treårig prøveprosjekt for innkjøpssamarbeid (LIS).
3.2.2.8 Reguleringer av detaljistvirksomheten/apotekdriften
Omsetning av legemidler er regulert i lov av 21. juni 1963 nr. 17 om drift av apotek (apotekloven), og i lov av 4. desember 1992 nr. 132 om legemidler mv. (legemiddelloven). Det er gitt en rekke forskrifter til nevnte lover. Det er også gitt instrukser i form av retningslinjer og rundskriv. Sosial-og helsedepartementets myndighet etter apotekloven er i det vesentlige delegert til Statens helsetilsyn.
Detaljistleddet særpreges av at apotekene omsetter spesialvarer som stiller krav til sikkerhet, leveringsdyktighet og kunnskapsformidling. Apotekene utfører også oppgaver som det offentlige har pålagt dem, såsom innkreving av egenandeler for Rikstrygdeverket, informasjonsvirksomhet om egenandelssystemet, kontroll med forbruk av legemidler med innhold av narkotiske stoffer, arkivering av A- og B-preparatresepter, samarbeid med fylkeslegen i misbrukssaker mv.
Apotekene er, med unntak av sykehusapotekene, drevet av selvstendig næringsdrivende. Det er imidlertid myndighetene som fastlegger de økonomiske rammebetingelser, fastsetter apotekenes beliggenhet og antall, tildeler bevillinger, utarbeider og følger opp lover og forskrifter. Bakgrunnen for at myndighetene styrer apotekvesenet på en slik måte, er i St.prp. nr. 1 1996-97 Sosial-og helsedepartementet (statsbudsjettet for 1997) angitt slik under kap. 751 Apotekvesenet:
oppnå kvalitet og sikkerhet i legemiddeldistribusjonen
sørge for god apotekdekning innenfor gitte økonomiske rammer
sikre god ressursutnyttelse
holde kostnadsnivået nede og unngå utilsiktede privatøkonomiske gevinster
gjennom tilsynsvirksomheten ønsker myndighetene dessuten å inspirere apotekene til virksomhet som bidrar til rasjonell legemiddelbruk i samfunnet
Hensynet til kvalitet og sikkerhet i legemiddeldistribusjonen er et av de sentrale hensyn bak gjeldende apotekordning. Hensynet har ifølge myndighetenes uttalte helsepolitiske målsettinger samme gjennomslagskraft i dag.
Apotekenes enerett
Lovverket gir apotekene enerett til salg av legemidler til publikum. Eneretten omfatter både reseptfrie og reseptpliktige legemidler.
Den enerett som dagens apotekvesen har til detaljomsetning av legemidler blir ikke berørt av EØS-avtalen. Det er opp til de nasjonale myndigheter å bestemme hva som skal komme inn under apotekenes enerett, samt eierstruktur og lokalisering av apotek.
Leveringsplikt
Apoteket skal, jf. apotekloven § 4, 27 og 28, så hurtig som mulig ekspedere de legemidler, forbindingssaker og sykepleieartikler som forlanges. Apoteket har også plikt til å holde tilstrekkelig varebeholdning og skal alltid ha beholdning av det som forskrives (apoteklovens § 34). Dette innebærer at apotekene har leveringsplikt for alle legemidler på det norske markedet. Dersom apoteket ikke har det legemidlet eller andre varer som forskrives, må apoteket skaffe dem så hurtig som mulig.
Ekspedisjonsrett (apotekloven § 20)
Legemidler på resept kan bare ekspederes av personer med ekspedisjonsrett. Slik ekspedisjonsrett har her i landet farmasøytiske kandidater (cand. pharm.) og reseptarer. Ekspedisjonsretten innebærer at vedkommende på selvstendig faglig grunnlag er ansvarlig for alle oppgaver og vurderinger knyttet til ekspedisjon av legemidler etter resept.
Farmasøytiske kandidater har 5 års universitetsutdannelse fra Universitetet i Oslo eller (etter hvert) Universitetet i Tromsø, mens reseptarene har 2 1/2 års utdannelse fra Reseptarutdanningen, Høgskolen i Oslo.
Ekspedisjonsretten følger automatisk av avlagt eksamen som cand. pharm. eller reseptar, og kan følgelig ikke fratas en person selv om denne på en eller annen måte skulle vise seg uskikket til arbeid i apotek eller annen farmasøytisk arbeidsplass (f.eks. som følge av stoffmisbruk).
Farmasøytiske kandidater er i Norge den eneste yrkesgruppen som oppfyller EUs krav til farmasøytkompetanse.
Apotekbevilling
I apotekloven § 7 heter det: «For å drive apotek kreves bevilling meddelt av Kongen». Myndigheten er videre delegert til Statens helsetilsyn.
Apotekbevilling gis personer med cand. pharm.-eksamen og som ellers oppfyller formelle krav fastsatt i apotekloven. Utover formelle krav baserer Statens helsetilsyn seg på egne retningslinjer av 3. desember 1992 for vurdering av søkere til bevilling. I retningslinjene presiseres blant annet at det skal legges vekt på lederkvalifikasjoner og at det ved viderebefordring (dvs. at en apoteker søker og får bevilling til nytt apotek) skal legges stor vekt på hvordan søkeren har drevet sitt apotek. Et rådgivende utvalg sammensatt av medlemmer som representerer Norges Apotekerforening og Norges Farmaceutiske Forening gir råd til Statens helsetilsyn med hensyn til hvem som bør tildeles bevilling i det enkelte tilfelle.
Apotekeren kan, i henhold til bestemmelsene i Pensjonsordningen for apoteketaten, inneha bevillingen til vedkommende må si den fra seg ved oppnådd aldersgrense, dvs. inntil fylte 70 år. I enkelte tilfeller gir apotekloven mulighet for å tilbakekalle en bevilling, blant annet dersom lokaler, innredning, utstyr eller varebeholdning holdes i så dårlig stand at forholdene må anses uforsvarlige, eller at innehaveren, tross advarsler, grovt overtrer eller forsømmer sine plikter eller når innehaveren må anses varig uskikket til å drive apoteket på en forsvarlig måte. Det anføres spesielt i apotekloven at apotekbevilling faller bort dersom det blir bestemt at apotekene skal drives av det offentlige.
I tillegg til de private apotekene, driver også staten og fylkeskommuner apotek. I disse tilfeller har staten/fylkeskommunen bevilling og apotekeren arbeider under vanlige statlige/fylkeskommunale lønns- og arbeidstidsbetingelser. Helsetilsynet foretar også ansettelse av apotekere ved offentlige apotek, med unntak av de to apotekene som staten selv eier. Sosial- og helsedepartementet står for ansettelse av disse apotekerne (fra 1995).
Opprettelse/nedleggelse/sammenslåing
Avgjørelser om opprettelse, nedleggelse, sammenslåing, flytting og statusendring av apotek foretas av Statens helsetilsyn. Sosial- og helsedepartementet er ankeinstans. På bakgrunn av en offentlig utredning om apotekenes organisasjonsform og funksjoner (NOU 1979: 26 Apotekdriftens organisasjon og funksjoner) utarbeidet Helsedirektoratet i 1984 en Landsplan for apotekdekning, der det ble lagt en strategi for fremtidig utvikling av apotekvesenet. Planen forelå i revidert form i 1990 og 2. reviderte utgave forelå våren 1995. Planen er retningsgivende og ikke bindende for Statens helsetilsyn.
Apotek opprettes og nedlegges i henhold til apotekloven når det er ønskelig eller påkrevd av allmenne hensyn. Tidligere ble apotek vanligvis opprettet etter initiativ av kommuner, uten at det forelå noen offisiell plan for dette arbeidet. Da den første landsplanen ble laget, ble det også laget kriterier for opprettelser av apotek (f.eks. befolkningsgrunnlag). Disse kriteriene har i løpet av de 10 årene som har gått endret seg. Bl.a. har myndighetene, på grunnlag av forsøksvirksomhet, gått inn for å etablere «enklere» apotektyper, som krever mindre investeringer, har lavere driftsutgifter og derfor kan drives med et mindre kundegrunnlag. Som en følge av dette har man i løpet av en 10-årsperiode vært i stand til å utvide apotekdekningen vesentlig. Den nye landsplanen legger opp til en fortsatt betydelig økning i antall apotek, særlig i utkantstrøk. For perioden 1995-2000 er det planlagt opprettelse av 54 nye apotek, de fleste som filialapotek (dvs. filialer under et hovedapotek hvor det er ansatt en bestyrer). Tre apotek er planlagt nedlagt i samme periode. Fylkesvis fordeling av nyopprettelsene fremgår av tabell 3.9.
Tabell 3.9 Planlagte apotekopprettelser 1995 - 2000, fylkesvis oversikt
| Fylke | Antall apotek i drift pr. 1995 | Innbyggere pr. apotek i drift i dag | Forslag opprettelser | Forslag nedleggelser |
| Østfold | 20 | 11.938 | 2 | 0 |
| Akershus | 25 | 17.188 | 8 | 0 |
| Oslo | 44 | 10.832 | 3 | 1 |
| Hedmark | 20 | 9.370 | 1 | 0 |
| Oppland | 22 | 8.332 | 1 | 0 |
| Buskerud | 20 | 11.356 | 5 | 0 |
| Vestfold | 15 | 13.460 | 4 | 1 |
| Telemark | 14 | 11.653 | 2 | 0 |
| Aust-Agder | 8 | 12.393 | 1 | 0 |
| Vest-Agder | 11 | 13.506 | 1 | 0 |
| Rogaland | 23 | 15.249 | 5 | 0 |
| Hordaland | 28 | 14.992 | 1 | 0 |
| Sogn og Fjordane | 13 | 8.275 | 1 | 0 |
| Møre og Romsdal | 21 | 11.418 | 3 | 0 |
| Sør-Trøndelag | 19 | 13.441 | 3 | 1 |
| Nord-Trøndelag | 8 | 15.965 | 7 | 0 |
| Nordland | 21 | 11.467 | 3 | 0 |
| Troms | 9 | 16.636 | 3 | 0 |
| Finnmark | 7 | 10.920 | 0 | 0 |
| Hele landet | 348 | 12.427 | 54 | 3 |
Kilde: Landsplan fra Apotekdekningen, revidert utgave 1995 - 2000
Tabell 3.9 baserer seg på apotek i drift pr. 31.12.94 og folketallet pr. 31.12.93.
Forslag til opprettelser og nedleggelser omfatter også landsplanens forslag til vurderinger i planperioden, men ikke vurderinger som foreslås gjennomført ved neste revisjon av landsplanen.
Opprettelsene foregår etter søknad fra den enkelte kommune, men landsplanen virker som en motivasjon til å søke. Det er imidlertid den private apoteker som må opprette apoteket, finne lokaler etc. Søknaden fremmes gjennom fylkeslegene. Statens helsetilsyn fatter vedtak etter at berørte parter er hørt. Statens helsetilsyn avgjør om apoteket skal opprettes som selvstendig apotek eller som filialapotek og i tilfelle hvilket apotek som skal være hovedapotek. Prosedyrene for opprettelse av apotek er i stor grad nedfelt i apotekloven (§ 1) og delvis i administrativ praksis. Det gis statsgaranti for lån til opprettelser av apotek. Ordningen med statsgarantier administreres av Statens helsetilsyn etter fastsatte retningslinjer. Stortinget setter rammer for total sum garantier og for årlige garantitilstillelser.
Nedleggelser av apotek skjer nesten bare i forbindelse med at en apotekbevilling er ledig. Forslag om nedleggelse blir sendt på høring til de samme instanser som ved opprettelser. Når nedleggelse er vedtatt, betaler staten kompensasjon (takst) fordi anlegget ikke blir solgt videre.
Siden 1984 er det opprettet ca. 85 apotek, de fleste som filialapotek, og det er lagt ned ca. 15. Det er opprettet nye apotek på mindre steder der det tidligere ikke har vært apotek og i drabantbyene rundt de større byene, mens nedleggelsene stort sett har foregått i sentrum av de større byene. I tillegg er flere apotek flyttet innenfor sitt dekningsområde eller sin kommune, og en rekke apotek har skiftet status, enten fra selvstendig apotek til filial eller fra filial til selvstendig apotek.
Fordi opprettelse av apotek krever et visst befolkningsgrunnlag og deler av landet har en spredt bosetning og vanskelige kommunikasjoner, er det etablert medisinutsalg (underlagt apotek) for å sikre folks tilgang på reseptfrie legemidler. Det er ca. 1 250 medisinutsalg fordelt på vel 200 apotek, hvorav enkelte har over 20 utsalg. Det er flest utsalg i Vest-Norge, Trøndelag og Nord-Norge. Utsalgenes størrelse varierer mye, fra en omsetning på mindre enn 1 000 kroner pr. år til over 1 mill. kroner pr. år. De største utsalgene er etablert i «egne lokaler», dvs. apoteket leier lokalene og personalet er ansatt av apoteket. De minste utsalgene er såkalte «medisinkister». Disse er på steder som er så små at det ikke finnes vanlige forretninger og hvor kommunikasjonene er så vanskelige at andre løsninger er uhensiktsmessige. Ca. 10 pst. av omsetningen målt i verdi av de reseptfrie legemidlene omsettes gjennom medisinutsalg.
Tabell 3.10 Utviklingen i apoteknettet 1987-1995 (Antall apotek ved utgangen av det enkelte år)
| Hoved- | Filial- | Offentlig | ||
| apotek | apotek | apotek | Sum | |
| 1987 | 266 | 32 | 16 | 314 |
| 1988 | 265 | 34 | 17 | 316 |
| 1989 | 264 | 37 | 18 | 319 |
| 1990 | 260 | 41 | 19 | 320 |
| 1991 | 257 | 47 | 22 | 326 |
| 1992 | 256 | 55 | 24 | 335 |
| 1993 | 254 | 66 | 25 | 345 |
| 1994 | 252 | 70 | 26 | 348 |
| 1995 | 250 | 78 | 27 | 355 |
Kilde: Norges Apotekerforening,1996.
Takst ved overtakelse
Når en apotekbevilling er ledig og skal overtas av ny innehaver, gjennomføres en takstforretning. Ifølge apotekloven § 14 og 15 har en påtroppende apoteker rett og plikt til å overta inventar, utstyr og varebeholdning i den utstrekning det er i tilfredsstillende brukbar stand, tilpasset apotekets behov og er nødvendig for driften av apoteket. Prisen fastsettes (av to takstmenn oppnevnt av partene) etter retningslinjer fastsatt av Helsetilsynet. Det er inntatt bestemmelser om skjønn eller voldgift dersom det ikke oppnås enighet mellom selger og kjøper.
Bakgrunnen for dagens takseringsregler er at man ønsker å sikre at apoteket kan drives videre uten avbrudd etter en overtakelse, og uten at ny innehaver (og derved apotekvesenet) skal påføres unødige kostnader gjennom nyinnkjøp.
Godkjenning av investeringer
Fordi staten garanterer for lån til overtakelser, nybygg og ombygging av apotekanlegg, har myndighetene behov for å kontrollere de investeringene som foretas i apoteklokaler og -innredning. Innehavere er pålagt (jf. apotekloven § 25) å fremlegge planer til godkjenning for departementet på forhånd. Lånegarantier gis ikke utover godkjent kostnadsoverslag. Dessuten forhåndsgodkjenner Statens helsetilsyn husleiekontrakter og investeringer for de apotek som mottar statlig driftstilskudd. Disse innehaverne blir pålagt å fremlegge driftsbudsjett for apoteket etter investering, og blir holdt ansvarlig i forhold til disse dersom overskridelser og feilbudsjetteringer medfører behov for driftsstøtte (jf. apotekloven § 25 og 26).
Apotekenes åpningstider
Inntil september 1995 har Statens helsetilsyn fastsatt maksimale åpningstider for apotekene. I rundskriv av 20. september 1995 har Statens helsetilsyn gitt nye rammer for apotekenes åpningstider. Gjeldende fra samme dato er det nå fastsatt minimums åpningstider for apotekene. Dette innebærer at apotekenes åpningstider lettere kan tilpasses åpningstider forøvrig i lokalsamfunnet og nye handlemønstre, slik at tilgjengeligheten til legemidler øker. Noen apotek har i tillegg vært pålagt åpningstid utover vanlig åpningstid, såkalt vakttjeneste. Slik vakttjeneste varierer fra døgnvakt (kun Jernbanetorvets apotek, Oslo), til vakt frem til kl. 23.00 (bl.a. i Bergen, Stavanger og Trondheim) til vakt noen timer lørdag og søndag.
Avgift på omsetning - Apotekavgift
Ifølge apotekloven § 38 skal den som har bevilling til å drive apotek betale avgift til statskassen. Fra 1992 ble avgiftsgrunnlaget endret fra avgift på all apotekomsetning til avgift bare på legemiddelomsetningen. Dette hadde sammenheng med ønsket om å øke apotekenes konkurranseevne i handelsvaremarkedet.
Avgiftsmidlene går i hovedsak til å dekke tilskuddsbehovet ved apotek som er berettiget til og som får innvilget driftsstøtte, fraktrefusjonsordningen og Pensjonsordningen for apoteketaten. Trinn og satser for avgiften fastsettes hvert år av Stortinget.
Avgiften regnes på apotekenhetens totalomsetning av legemidler, fratrukket salg til sykehus og andre fylkeskommunale institusjoner. Apotekavgiften ble tidligere brukt til å utjevne apotekenes driftsresultater. Mens 75 pst. (33 mill. kroner) av en beregnet total avgiftsinngang på 44 mill. kroner var tenkt benyttet til tilskudd til apotek i 1992, er det for statsbudsjettet for 1997 anslått ilignet 69,5 mill. kroner, hvorav 22 pst., eller 15,5 mill. kroner, skal benyttes til tilskudd til apotek. Utviklingen har gått i retning av at apotekavgiftens betydning som utjevningsfaktor er blitt redusert.
Boks 3.1 Trinn og prosentsatser for beregning av apotekavgift for 1997
av de første 12.000.000 ingen avgift
av de neste 8.000.000 2,5 pst.
av de neste 10.000.000 3 pst.
av resten 4 pst.
det gis et bunnfradrag på 6 mill. kroner for hvert filalapotek i avgiftsgrunnlaget
Med disse trinn og satser er avgiften beregnet å innbringe 69,5 mill. kroner, og skal bl.a. dekke følgende utgifter:
tilskudd for Pensjonsordningen for apoteketaten 7,0 mill. kroner
legemiddelstatistikk 3,0 mill. kroner
tilskudd til apotek 15,5 mill. kroner
stipendier 0,5 mill. kroner
tilskudd til fraktrefusjon av legemidler 36,1 mill. kroner
tilskudd til institutt for farmakoterapi 3,3 mill. kroner
tilskudd til regionale legemiddelsentra 6,1 mill. kroner
Apotekavansen
Apotekets avanse fastsettes av sentrale helsemyndigheter. Avansen kan justeres fortløpende for å holde en rimelig gjennomsnittlig inntjening. Denne vurderingen blir foretatt flere ganger i året. Det blir i denne forbindelse utarbeidet samlede prognoser for de private apotek basert på de siste års regnskap korrigert for forventede endringer i pris- og omsetningsutvikling, lønnsforhold og andre forventede kostnadsendringer. Forventet behov for statlig støtte beregnes.
Fra 1. september 1992 er det innført en klart mer degressiv avanse enn tidligere. Et nytt avansesystem ble innført fra 1. januar 1995, der hensikten er at den enkelte legemiddelpakning i større grad skal bære de kostnader som er forbundet med apotekets håndteringskostnader. For å oppnå dette er strukturen i avansen lagt om til større fast kronetillegg (uavhengig av innkjøpspris) og lavere prosenttillegg (med utgangspunkt i innkjøpspris). Omleggingen var et ledd i arbeidet med å innføre mer «kostnadsriktige avanser» på grossist- og detaljistleddet, og vil bl.a. resultere i at det samlede driftsresultat for apoteksektoren vil bli redusert. Som en videreføring av dette arbeidet, ble det fra 1. september 1995 iverksatt en ny avanseendring, hvor apotekavansen i hovedsak ble redusert på de dyreste preparatene.
De siste årene er det foretatt mange avansereduksjoner, senest 1. september 1996. Det er vedtatt en ny avansenedsettelse fra 1. februar 1997.
Tildeling av driftstøtte
Fra 1. januar 1994 er det innført et nytt støttesystem for apotekvesenet. Hovedelementene er her at apotekene i utgangspunktet skal være økonomisk uavhengige. Statens støtte er øremerket apotek med liten omsetning og som er beliggende i utkant-Norge (begge vilkår må være oppfylt). Hensikten er å sikre tilgjengelighet i de deler av landet der betingelsene for apotekdrift ikke fullt ut er tilstede. Videre vil apotek som utfører bestemte samfunnsfunksjoner kunne motta driftstøtte. Støtte kan gis som ettergitt apotekavgift eller som ren driftstøtte. Det vises til kap. 8.4.8.5 for nærmere omtale av ordningen. Tabellen viser utviklingen av driftstøtten til apotekene i perioden 1988-94.
Tabell 3.11 Driftstøtte til apotek 1988-94, løpende kroner.
| Ettergitt avgift (mill. kroner) | Tilskudd (mill. kroner) | Antall støtte- apotek | Netto omsetning private apotek (mill. kroner) | |
| 1988 | 12,9 | 45,1 | 126 | 3.371 |
| 1990 | 10,4 | 22,7 | 75 | 4.129 |
| 1992 | 2,3 | 18,6 | 52 | 4.922 |
| 1993 | 3,2 | 12,4 | 41 | 5.333 |
| 1994 | 1,6 | 6,1 | 27 | 5.690 |
| 1995 | 0,7 | 5,0 | 17 | 6.257 |
Kilde: Statens helsetilsyn
Utviklingen i utbetaling av driftstøtte til apotek i denne perioden må bl.a. ses i sammenheng med utviklingen i apotekenes økonomi (se kapittel 8.2).
Fraktrefusjonsordningen
Fraktrefusjonsordningen er beregnet på pasienter som har vanskelig for å oppsøke apotek. Ordningen bidrar til at reseptpliktige legemidler ikke blir dyrere for disse pasientene.
Ordningen virker ved at apotekenes fraktutgifter ved nødvendig forsendelse av reseptforskrevet medisin refunderes av det offentlige. Ordningen ble til og med 1992 administrert av Norsk Medisinaldepot (NMD), og kostnadene ble dekket av NMDs avanse. Fra 1993 har Norges Apotekerforening administrert ordningen i henhold til avtale med Statens helsetilsyn. Fra samme tidspunkt er ordningen finansiert av apotekavgiften. Retningslinjene for refusjon fastsettes av Statens helsetilsyn.
I 1995 ble det refundert frakt for i alt 1,8 millioner pakker, noe som innebærer at ca. 10 pst. av alle utstedte resepter ekspederes ved forsendelse. Ordningens totale kostnader var 36 mill. kroner. Antallet refunderte pakker viste en vekst på nærmere 5 pst. i forhold til 1994, mens ordningens totale kostnader lå på samme nivå.
Det er forutsatt at forsendelsene skal skje på en kostnadseffektiv måte. Ordningen skal ikke benyttes av apotekene som ordinær service eller i salgsfremmende øyemed overfor pasienter/kunder. Det er bevilget 36,1 mill. kroner til dette formålet i 1997.
3.2.2.9 Endringer av offentlige reguleringer gjennomført på legemiddelområdet de senere år (nærmere omtalt på ulike steder i rapporten)
Boks 3.2 Endringer av offentlige reguleringer
fra 1.1.89
innføring av prosentvis egenandel på legemidler refundert av folketrygden
fra 1.4.91
krav til å forskrive billigste synonympreparat
fra 1.9.93
referanseprissystem for generiske legemidler
fra 1.1.94
omlegging av driftstøttesystemet for apotek
opphevelse av behovsparagrafen for godkjenning av legemidler fra 1.1.95
fri prisdannelse på reseptfrie medisiner
opphevet statlig regulering av prisene på produsentleddet (GIP)
opphevelse av målsetting om lik pris på landsbasis, men lik maksimalpris
mer kostnadsriktige avanser i apotekvesenet og NMD
direkte leveranser av legemidler fra grossist/produsent til sykehus for utvalgte legemiddelgrupper som sykehusene er storforbrukere av. Innkjøpene ble forutsatt gjennomført etter anbudsprinsippet
nye helseøkonomiske prosedyrer, større vekt på økonomisk evaluering av legemidler som et viktig beslutningsgrunnlag i pris- og refusjonspolitikken
NMDs enerett til import (eksport) og engrosomsetning opphevet
adgang til parallellimport
3.3 Dagens refusjonssystem for legemidler
3.3.1 Blåreseptordningen
Historisk oversikt
Det er 43 år siden det ble inntatt allmenne bestemmelser i trygdelovgivningen om pliktmessig dekning av utgifter til livsviktige legemidler. Ordningen trådte i kraft høsten 1953, og besto i en prosentvis refusjon av utgifter utover et minstebeløp på 50 kroner. For enkelte sykdommer var dekningen 100 prosent over minstebeløpet, mens det for de fleste gjaldt en refusjon på 75 prosent uten noen øvre grense for egenbetalingen. Som hovedregel måtte behandlingen være instituert på sykehus eller av spesialist. Det var utarbeidet en liste over kroniske sykdommer og en liste med kostbare legemidler i forskriftene om den første blåreseptordningen.
Tidligere var det bare visse yrkesbetingede sykdommer som ga rett til trygdemessig dekning av legemiddelutgifter, men det hadde vokst frem en del skjønnsmessige bidragsordninger forut for lovgivningen i 1953. I tiden etter har det funnet sted hyppige regelendringer og justeringer.
Forutsatt at visse betingelser for en sykdoms alvorlighetsgrad og varighet var oppfylt, gjaldt det fra sommeren 1960 og ut 1980 at trygden dekket alle legemidler. Samtidig måtte alle øvrige legemidler privatfinansieres fullt ut (om de ikke unntaksvis kom inn under en bidragsordning eller spesielle lovbestemmelser). Ordningen med enten full offentlig eller full privat betaling kunne gi inntrykk av at det finnes et absolutt skille mellom (livs)nødvendig medikasjon og annen medikasjon.
I 1966 ble reglene som begrenset adgangen til refusjonsberettighet behandling oppmyket slik at ikke-spesialister fikk hånd om en langt større del av forskrivningen. Samtidig ble en samlegruppe av sjeldne kroniske tilstander (innført i 1960) utvidet til å gjelde også vanligere lidelser, og det ble innskjerpet at rett til trygdedekning først inntrådte når en sykdom var gått inn i en kronisk fase. Omtrent på samme tid ble også billigere preparater inntatt på listen. Det offentlige overtok på denne måten så godt som alle utgifter til medikasjon for et stort utvalg av langvarige lidelser.
Fra 1981 ble det igjen innført et begrenset privatøkonomisk delansvar med en fast egenandel på 40 kroner for voksne. Barn og pensjonister ble unntatt i første omgang, men fra 1984 ble det innført halv egenandel for disse, tilsvarende 25 kroner, mens egenandelen for voksne økte fra 40 til 50 kroner (se tabell 3.12 for utviklingen av egenbetalingsnivået i Norge).
Gjeldende regler for refusjon
Folketrygdloven kap. 2, § 02-05 nr. 3 bokstav a, hjemler godtgjørelse av utgifter til viktigere legemidler etter forskrifter som fastsettes av Sosial- og helsedepartementet. Nærmere regler for refusjon av utgifter til legemidler fremgår av «Forskrifter om godtgjørelse av utgifter til viktigere legemidler».
Utgifter til viktigere legemidler og sykepleieartikler refunderes ved behandling av nærmere angitte sykdommer oppført i forskriftenes § 9. Forskriftene angir også hvilke hovedgrupper av legemidler det kan gis refusjon for. I tilhørende preparatliste er de legemidler som kan refunderes ved de enkelte sykdommer listet opp. Legemidlene skal være forskrevet av lege (allmennlege eller spesialist), eller behandlingen skal være instituert i sykehus eller hos spesialist.
Hovedregelen for å få refusjon er at sykdommen er gått inn i en langvarig fase, og at det er behov for langvarig medikamentell behandling i minst 3 måneder i løpet av et år, enten sammenhengende eller i form av gjentatte, kortere kurer for den samme sykdommen. Dersom hovedvilkårene er oppfylt, kan legemidlene på listen forskrives på blå resept og ekspederes fra apotek uten spesiell godkjenning fra trygdekontoret. For at et preparat skal bli tatt opp på preparatlisten til § 9, skal legemidlet som hovedregel være godkjent av Statens legemiddelkontroll for bruk ved en av de sykdommer som er nevnt i den aktuelle paragraf, og inngå i en av de legemiddelgrupper som er nevnt i forbindelse med den enkelte sykdom i § 9.
Ved sykdom som ikke er nevnt i forskriftenes § 9, kan det unntaksvis ytes godtgjørelse etter forskriftenes § 2. Unntaksvis tolkes i praksis slik at det må dreie seg om en sjelden sykdom eller et behandlingsalternativ som sjelden er aktuelt. Refusjon etter denne bestemmelsen ytes etter individuell søknad til trygdekontoret. Det samme gjelder for forskrivning av legemiddel som ikke er nevnt i preparatlisten til forskriftenes § 9 ved en av de sykdommer som er nevnt i denne.
For preparater som ikke er ført opp på blåreseptlisten kan det i enkelte tilfeller (etter individuell søknad) ytes refusjon etter forskriftenes § 10.2. På denne måten kan man sette bestemte vilkår for refusjon og lettere sikre at de vilkår man setter for refusjon er oppfylt. Dette har i flere tilfeller vært gjort i den første tiden etter at et legemiddel er kommet på markedet. Når man etter en tid har vunnet erfaring med preparatet, er det blitt oppført på blåreseptlisten.
Kortvarige kurer dekkes ikke etter disse forskrifter, selv ikke for meget kostbare legemidler. Det er gjort unntak for allmennfarlige smittsomme sykdommer.
Det er helsemyndighetene som, ut fra faglig vurdering og vurdering av helseøkonomisk effekt ved de ulike terapiformer, avgjør om det offentlige skal gi pasienten rett til å få dekket utgiftene som knytter seg til et spesifikt preparat.
Sosial- og helsedepartementet har nedsatt et utvalg som skal vurdere prinsippene for trygderefusjon av legemidler. Utvalget vil komme med sine tilrådinger i begynnelsen av 1997.
3.3.2 Egenbetaling
Egenandelen er fra 1. juli 1992 fastsatt til 30 pst. av reseptbeløpet, maksimalt 300 kroner. For barn under 16 år og alders- og uførepensjonister er egenandelen 10 pst. av reseptbeløpet, maksimalt 75 kroner. For barn under 7 år betales ikke egenandel.
Egenbetalingsordningen har gjennomgått en del endringer i løpet av de år folketrygden har refundert utgifter til medisiner for pasienter med fortrinnsvis kroniske lidelser. Fra og med innføringen av prosentvis egenandel i 1989, har det vært en viss justering av pasientenes egenandeler, jf. tabellen under. Begrunnelsen for å la egenbetalingen avspeile kostnadene var at dette i større grad skulle stimulere til nøkternhet i forskrivningen, både i valget mellom ulike legemidler med forskjellig pris, og i valg av forskrevet mengde (særlig utprøving av legemidler ved oppstart av behandling). Dette skulle derfor bidra til å redusere svinn og begrense trygdens utgifter.
Tabell 3.12 Utvikling av egenbetalingsnivået i Norge
| År | Egenbetaling | Maksimalt pr. resept (kroner) | Redusert egenbetaling* | Maksimalt pr. resept (kroner) |
| 1988 | 60 kroner pr. resept | 30 kroner pr. resept | ||
| 1989 | 20 pst | 150 | 10 pst | 75 |
| 1990 | 20 pst | 175 | 10 pst | 75 |
| 01.07.92 | 30 pst | 300 | 10 pst | 75 |
| 01.01.97 | 36 pst | 330 | 12 pst | 110 |
* For barn mellom 7 år og 16 år og alders- og uførepensjonister.
3.3.3 Skjermingsordningen
I 1984 ble ordningen med et felles årlig utgiftstak for egenandeler innført. Hensikten var å skjerme storforbrukerne av helsetjenester for urimelige totalutgifter. Skjermingsordningen omfatter konsultasjon hos lege eller klinisk psykolog, reiseutgifter, røntgenundersøkelse og legemidler på blå resept. Egenbetaling ved de tjenester som inngår i ordningen kvitteres på et spesielt kort utsendt av Rikstrygdeverket. Når samlet egenbetaling fra årets begynnelse når egenandelstaket innløses kvitteringskortet i frikort, og brukeren belastes ikke egenandeler ut kalenderåret. For barn under 16 år beregnes utgiftene samlet for alle søsken og en av foreldrene. Egenandelstaket er satt til 1 290 kroner for 1997. Statens utgifter til dekning av egenandeler for legemidler utover egenandelstaket var på 141,1 mill. kroner i 1995.
3.3.4 Bidragsordningen - § 2-13
Kostbare, reseptpliktige legemidler som ikke dekkes pliktmessig etter ovennevnte forskrifter, kan dekkes delvis ved bidrag etter folketrygdelovens § 2-13. Når det gjelder store faktiske utgifter for den enkelte, kan det i dag søkes om bidrag for dekning av 2/3 av de utgifter som overstiger 600 kroner. Pasienter som har kortvarige lidelser, men hvor kostnadene er store, kan derved søke om å få disse delvis dekket over folketrygdens bidragsordning. Utgifter til flere legemidler kan ses under ett i forbindelse med en slik søknad. I forhold til utgiftene over refusjonsordningen er trygdens utgifter til bidrag relativt begrensede. I 1995 beløp disse utgiftene seg til 78 mill. kroner.
3.3.5 Grunnstønad
Grunnstønad skal, helt eller delvis, kompensere ekstrautgifter av betydning som er forårsaket av varig sykdom, skade eller lyte. Det er tale om utgifter som følger direkte av den medisinske tilstanden som f.eks. ekstra transportutgifter, drift av tekniske hjelpemidler o.l. Det kan også gis grunnstønad til mer behandlingspregede utgifter som ikke dekkes pliktmessig. Grunnstønad er ment å være en varig ytelse og vil som oftest løpe i årevis eller livet ut.
Når den trygdedes medisinutgifter dekkes etter blåreseptordningen (§ 2-5), gir ikke disse utgiftene rett til grunnstønad. Det gis heller ikke grunnstønad til dekning av egenandeler. Som nevnt i punktet over, kan det i enkelte tilfeller gis bidrag for å dekke visse utgifter til medisiner etter § 2-13 første ledd. Når vilkårene for rett til grunnstønad er oppfylt (bl.a. varighet og utgiftsbeløp), skal grunnstønad, og ikke § 2-13, anvendes.
3.3.6 Krav om forskrivning av billigste synonympreparat
Norske leger ble fra 22. april 1987 oppfordret til å forskrive det billigste legemidlet hvis det fantes flere likeverdige alternativer. Oppfordringen ble erstattet med et påbud om å forskrive billigste synonympreparat fra 1. april 1991.
Synonympreparater er preparater med samme virksomme stoff i samme mengde og samme legemiddelform (eks. tabletter, mikstur osv.), og som av Statens legemiddelkontroll er vurdert som terapeutisk (medisinsk) likeverdige. Prisene på generiske preparater kan variere betraktelig. Vanligvis vil originalpreparatet være dyrere enn de synonympreparatene som er kommet til etter at patentet på originalpreparatet er utløpt. For en del legemiddelgrupper har originalpreparatet likevel vært det dominerende på markedet. De legemidler som ble omfattet av ordningen med billigste synonympreparat var stort sett store legemiddelgrupper til behandling av bl.a. hjerte-karsykdommer, allergi, magesår og muskel-skjelettlidelser.
Innenfor flere legemiddelgrupper med stor omsetning og mange generiske preparater har man sett en endring i forbruksmønsteret, fra vesentlig forskrivning av originalpreparat til stadig større bruk av billigere synonympreparater. Markedsandelen for billigste generiske preparat målt i omsetning var ca. 34 pst. ved inngangen til 1991. Andelen økte til 42 pst. ved inngangen til 1992, og til 52 pst. ved inngangen til 1993. Grovt anslått medførte økningen en besparelse på 20 mill. kroner i 1992 (NMDs årsmelding, 1992). Totalt utgjør legemiddelgruppene som berøres av regelen om billigste synonym vel 10 pst. av totalomsetningen i kroner av registrerte legemidler.
3.3.7 Referanseprissystemet
Ordningen med pliktmessig forskrivning av billigste synonympreparat førte ikke til at veksten i det offentliges samlede legemiddelutgifter ble vesentlig bremset opp. 1. september 1993 ble referanseprissystemet innført. Dette systemet bygger på det samme prinsipp som ordningen med billigste synonympreparat, og innebærer at det fastsettes en maksimal pris for hva trygden pliktmessig skal refundere for en gitt gruppe generiske preparater. Denne maksimalprisen, kalt referanseprisen, er hittil fastsatt som prisen på billigste synonym i gruppen + 5 pst. Egenandelen beregnes av referanseprisen.
Dersom legen forskriver et preparat med høyere pris enn referanseprisen, må pasienten betale ordinær egenandel, tilsvarende 30 pst. av referanseprisen. I tillegg må pasienten betale forskjellen mellom referanseprisen og utsalgsprisen på det forskrevne preparatet. Den overskytende del vil ikke telle med under det totale egenandelstaket (i skjermingsordningen).
Ordningen omfatter 7 legemiddelgrupper, med til sammen 475 preparater. Omsetningen av disse preparater var ca. 700 mill. kroner (AUP) på årsbasis i 1993. Ordningen førte raskt til at så godt som alle preparater i en gruppe fikk samme pris, svarende til referanseprisen. Legemiddelindustriforeningen har med utgangspunkt i oppgaver fra medlemsbedrifter beregnet den årlige besparelse til 115 mill kroner.
Referanseprislisten er ajourført en gang (i 1995) siden referanseprissystemet ble innført.