3 Arv og kreft – en virkelighetsbeskrivelse
3.1 Det norske kreftbildet
3.1.1 Begrepene insidens og prevalens
Krefthyppighet, eller insidens, blir angitt som antall nye krefttilfeller i løpet av et kalenderår i forhold til en befolkningsmengde; vanligvis per 100.000. Ofte presenteres dette som alderskorrigert insidens, gjerne med verdensbefolkningens alderssammensetning som referansestandard.
Prevalens angir egentlig antallet personer som på et bestemt tidspunkt har en sykdom. Ved kreftsykdommer forstås begrepet prevalens som antallet personer som en gang har fått en kreftdiagnose og som er i live, enten helbredet eller fortsatt syke.
3.1.2 Sykdomshyppighet (insidens)
I 1996 fikk totalt 20.780 personer kreft i Norge; 10.229 kvinner og 10.551 menn. 1 Brystkreft er den hyppigste kreftform hos kvinner og utgjør 12% av all kreft og 24% av all kreft hos kvinner. Kreft i prostata (blærehalskjertelen) er den hyppigste kreftform hos menn og utgjør 12% av all kreft og 23% av all kreft hos menn. Én av ti menn får kreft i prostata og én av tretten kvinner kreft i brystet.
Kreft i tykk- og endetarm oppstår hyppig både hos kvinner og menn; disse to kreftformene utgjorde i 1996 til sammen 1.531 av de nye krefttilfellene blant kvinner og 1.379 blant menn. Også lungekreft er en av de vanligste kreftformene; i 1996 var det 659 nye lungekrefttilfeller blant kvinner og 1.243 blant menn.
Kreft i fordøyelsesorganene omfatter kreft i spiserør, magesekk, tynntarm, tykk- og endetarm, lever, galleveier og bukspyttkjertel (pancreas) og utgjør til sammen ca. 25% av alle krefttilfeller.
7,2% av alle krefttilfeller er underlivskreft hos kvinner – det vil si kreft i eggstokk, livmorlegeme, livmorhals eller kjønnsorganer. Dette utgjør 14,7% av all kreft hos kvinner.
Kreft er en sjelden tilstand hos barn. Kreft hos barn utgjør 0,7% av all kreft (153 nye tilfeller i 1996). Ca. 10 tilfeller per år er arvelige kreftsykdommer.
Omkring 90% av alle krefttilfeller oppstår etter 50-årsalderen.
En gutt som fødes i dag vil ha en risiko på 2,9% for å få kreft før han er 50 år og 32,3% før han er 80 år. Dette er under forutsetning av at risikoen for å få kreft forblir hva den er i dag.
Legger man dagens prosentandeler til grunn og predikerer for de kommende 90 år, vil omlag 1,7 millioner nordmenn få kreft i dette tidsrommet. Av disse vil 160.000 være lungekreftpasienter og 170.000 brystkreftpasienter. Rundt 40% av landets innbyggere på 4,3 millioner vil i løpet av sin levetid utvikle kreft.
3.1.3 Hvor mange lever med sykdom (prevalens)
Det er et stort antall mennesker i live i Norge som har gjennomgått kreftdiagnostikk og -behandling. I hele landet dreide det seg om ca. 130.000 personer ved utgangen av 1996. Blant disse er det særlig de 55.000 kreftpasientene som fikk diagnosen nylig (for mellom 0 og 5 år siden) som representerer behandlingsutfordringene. Ved utgangen av 1995 fantes det ca. 1950 personer i live i Norge som hadde fått en kreftdiagnose før de fylte 15 år.
3.1.4 Forventet utvikling fram mot år 2010
Antallet krefttilfeller er avhengig av befolkningens størrelse, alderssammensetningen og risikoen for å få kreft. Siden kreft særlig rammer eldre, vil antallet krefttilfeller øke med antallet eldre i samfunnet.
Kreftforekomsten øker i Norge, og fra 1985 til 2010 er det beregnet en økning i den årlige forekomsten av kreft på 2.576 tilfeller hos menn (30,6% økning) og 2.918 (37,7% økning) hos kvinner, til sammen 5.494 tilfeller. Vi regner med at rundt 45% av økningen skyldes endringer i befolkningens alderssammensetning og de resterende 55% økt risiko for å få kreft.
I samme periode, fra 1985 til 2010, er det beregnet at antall kreftdødsfall per år kommer til å øke med 1.317 (25,9%) blant menn og 1.321 (31,2%) blant kvinner, til sammen 2.638 flere kreftdødsfall i år 2010 sammenliknet med 1985.
Fra 1993 til 2010 vil vi oppleve en økning på 18,7% nye krefttilfeller, samtidig som antall kreftdødsfall stiger med 13,6%.
3.1.5 Tilfeller av arvelig kreft
Vi kjenner ikke de nøyaktige tallene, men hvis vi antar at ca. 5% av tilfellene av brystkreft, prostatakreft, eggstokkreft og tykk- og endetarmkreft er arvelig betinget, betyr det at ca. 400 av kreftdiagnosene i 1996 dreide seg om arvelig kreft. De arvelige krefttypene har vanligvis en lavere debutalder enn de tilsvarende ikke-arvelige typene. Det er grunn til å tro at leveutsiktene for pasienter med arvelig kreft, er minst like gode som for den totale kreftpopulasjonen. Det betyr at prevalenstallet er omkring 2.000, men det kan være atskillig høyere fordi lavere alder ved diagnose kan ha medført bedrede leveutsikter.
Det er en liten prosentandel av det totale antall krefttilfeller som skyldes en nedarvet dominant disposisjon (se punkt 3.2.1). Det store antall, i størrelsesorden 90%, er sporadiske tilfeller. Imidlertid finnes det en gråsone-gruppe av ukjent størrelse som har en opphoping av kreft i familien (familiære tilfeller) hvor betydningen av de arvelige disposisjonene ikke er kjent. Dessuten finnes det flere sjeldne syndromer der flere ulike typer kreft kan være en del av sykdomsbildet. Figur 3.1 og 3.2 eksemplifiserer denne inndelingen for to av de fire store kreftsykdommer i den vestlige verden, henholdsvis bryst- og eggstokkreft og tykk- og endetarmkreft.
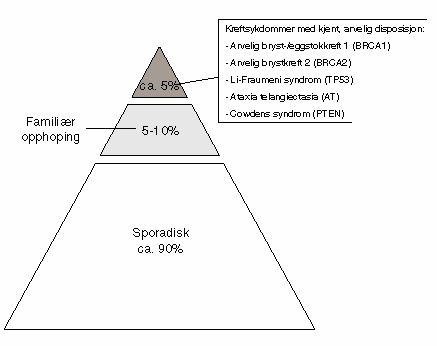
Figur 3.1 Fordelingen av bryst- og eggstokkreft mellom tilfeller med kjent arvelig disposisjon, familiær opphoping og sporadiske tilfeller. Gener som i endret tilstand disponerer for sykdommen står i parentes.
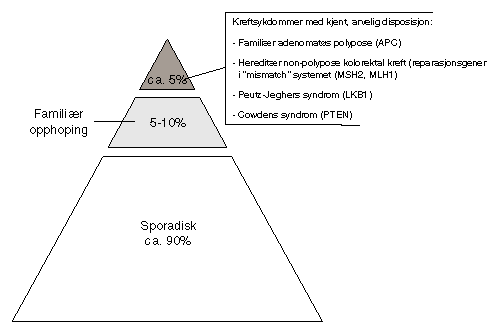
Figur 3.2 Fordelingen av tykk- og endetarmkreft mellom tilfeller med kjent arvelig disposisjon, familiær opphoping og sporadiske tilfeller. Gener som i endret tilstand disponerer for sykdommen står i parentes.
3.2 Kreft, arv og gener
3.2.1 Arvemønstre
Gener er deler av arvestoffet (genomet), som er bygget opp av DNA. Arvestoffet er organisert i 46 kromosomer – 22 par autosomer (kromosomer som ikke er kjønnskromosomer) og 2 kjønnskromosomer. Genene foreligger derfor parvis, på hvert sitt kromosom. Det ene genet i hvert par er arvet fra far, det andre fra mor.
Sykdommer følger ulike arvemønstre. Ved enkel arv, når bare ett par gener er involvert, snakker man om autosomal eller kjønnsbundet og dominant eller recessiv arv. Autosomal arv innebærer at genparet sitter på et av de 22 par autosomer; ved kjønnsbundet arv følger sykdommen et av kjønnskromosomene (X, Y). Ved dominant arv er det tilstrekkelig at ett gen inneholder en feil; ved recessiv arv må begge gener i et genpar inneholde feil for å gi sykdom.
Ved arvelig kreft er autosomal dominant arv det vanlige mønsteret. Ved dette arvemønsteret har barn av syke 50% risiko for å arve sykdomsgenet. Alle som har sykdomsgenet får sykdommen. Friske personer har ikke genfeil. Det er mange unntak fra disse reglene, men viktigst i denne sammenhengen er nedsatt penetranse (se nedenfor).
I tillegg til enkel arv, kan sykdommer arves multifaktorielt, og dette gjelder de fleste alminnelige sykdommer. Slike sykdommer har sin årsak i et samspill mellom flere genpar og miljøfaktorer. Dette gjelder også de fleste alminnelige kreftsykdommene.
3.2.2 Kreft og genfeil
Med mutasjon (genfeil) menes i vid forstand en permanent endring i basesekvensen i et gen. Mange slike endringer er en del av den normale variasjonen, og har ingen sammenheng med sykdom. Derfor brukes begrepet mutasjon ofte i en mer snever betydning: om en endring som henger sammen med sykdom. Det er i denne betydningen begrepet brukes her. Til praktiske formål i medisinen kan begrepet brukes enda snevrere: om endring som gir en høy risiko for sykdom, for eksempel minst 25% livstidsrisiko for sykdom.
For at en kreftsvulst skal etableres og utvikles, er en oppsamling av flere genfeil nødvendige. I denne forstand ligger genfeil til grunn for all kreft. Genfeilene finnes som regel bare i svulsten. Men i noen tilfeller arves en mutasjon i et kreftgen. Mutasjonen vil da finnes i alle celler i kroppen. Individer med en slik mutasjon i alle celler, er arvelig disponert for å få kreft.
3.2.3 Arvelig disposisjon for kreft
Når arvede mutasjoner (kimbanemutasjoner) i kreftgener har høy penetrans (høy sannsynlighet for å føre til sykdom), vil kreftsykdommen følge et dominant arvemønster i familien. For at en kreftsvulst skal etableres, er det i tillegg til den arvede genfeilen nødvendig med somatiske mutasjoner (genfeil i cellen som blir opphavet til kreftcellene). Derfor debuterer også arvelig kreft som regel først i voksen alder, men likevel tidligere enn sporadisk (ikke-arvelig) kreft.
En dominant arvelig disposisjon medfører også en økt risiko for tosidig kreft i parede organer som bryst, eggstokker, øyne eller nyrer.
3.2.4 Gener som disponerer for kreft
Det er to hovedtyper av gener som er direkte medvirkende til kreft: tumor-suppressorgener og onkogener. I tillegg kan skader i såkalte reparasjonsgener indirekte forårsake kreft ved å medvirke til at kreftrelaterte gener ikke opprettholder sin normale funksjon.
3.2.4.1 Tumor-suppressorgener
Tumor-suppressorgener (også omtalt som bremsegener) koder for proteiner som normalt har en veksthemmende funksjon i cellen. Feil i slike gener kan føre til mangel på proteindannelse eller produksjon av et endret protein, og dermed også en mulighet for ukontrollert vekst. Blant de tumor-supressorgener vi kjenner til, er mange assosiert med arvelige kreftsykdommer.
Et eksempel er retinoblastomgenet ( RB1). Retinoblastom er den hyppigste kreftform i øyet hos barn. I 20-30% av tilfellene manifesteres sykdommen tosidig, det vil si at begge øyne er angrepet. Disse tilfellene, og ca. 15% av de ensidige tilfellene, følger dominant arvegang. På cellenivå opptrer genet likevel recessivt: begge genkopier må være muterte for at genet skal miste sin normale funksjon. Individer med den arvelige formen har allerede én mutasjon i alle sin retinaceller, så en somatisk RB1-mutasjon i en tilfeldig retinacelle vil inaktivere genet i denne cellen og alle dens avkom. Dermed kan en kreftsvulst oppstå. I sporadiske (ikke-arvelige) tilfeller må to somatiske mutasjoner (i begge kopier av genet) ramme samme cellelinje for å resultere i en tilsvarende inaktivering.
3.2.4.2 Onkogener
Mens tumor-suppressorgener normalt har en veksthemmende virkning, koder proto-onkogener for proteiner som virker stimulerende på celledeling. Disse genene inngår også ofte som komponenter i cellens signaloverføringer. Onkogener er muterte utgaver av de normale proto-onkogener. Et onkoprotein (proteinproduktet fra et onkogen) kan ha sterkt økt aktivitet i forhold til det normale proteinet, enten fordi det ikke lar seg påvirke av cellens reguleringsmekanismer, eller fordi det produseres for mye av det (det overuttrykkes).
Så langt vi vet er onkogener sjelden nedarvet, og de har derfor liten betydning for arvelig kreft. Blant de eksempler vi kjenner er MET-onkogenet som disponerer for papillær nyrekreft.
3.2.4.3 Reparasjonsgener
Mutasjoner i tumor-suppressorgener og i protoonkogener er direkte årsak eller bidrag til at kreft utvikles. Mutasjoner i en eller flere komponenter i cellens reparasjonssystemer kan indirekte bidra til kreftutvikling, ved at skader som oppstår i andre gener ikke blir reparert. Hvis et tilstrekkelig antall sentrale, vekstregulerende gener skades, kan dette føre til ukontrollert vekst og etablering og utvikling av kreft.
Reparasjonsgener har det til felles med tumor-supressorgener at begge genkopier må ha feil for at funksjonen skal være tapt.
Den sjeldne, arvelige sykdommen xeroderma pigmentosum er et eksempel på nedsatt evne til å reparere skader i DNA. Den genetiske feilen bak kolorektal/livmorkreft (HNPCC) er også feil i et DNA reparasjonssystem.
3.2.4.4 Gener involvert i omsetning av karsinogener
Visse gener har betydning for utvikling av kreft fordi ulike varianter av dem omsetter karsinogener (kreftframkallende stoffer) med ulik effektivitet. Røykere som bryter ned stoffet debrisoquine raskt, har for eksempel en tilleggsrisiko for å utvikle lungekreft, sammenliknet med røykere som omsetter stoffet langsommere. Det finnes mange slike detoksifiseringsgener som virker på liknende måte.
3.2.5 Kromosomforandringer
Kromosomforandringer som kan sees i lysmikroskop, er vanlige i kreftsvulster. Undersøkelse av disse kan være nyttige til differensialdiagnostikk og er dermed med på å bestemme behandling. I noen tilfeller har slike funn også prognostisk verdi.
3.3 Tilbudet til risikopersoner i dag
Helsevesenets primære hensikt med å tilby et kontrollopplegg for personer med høy kreftrisiko, er å redusere dødeligheten av kreft ved tidlig påvisning og behandling av sykdommen. Det er også et mål å gi personer med opphoping av kreft i familien tilstrekkelig informasjon, blant annet om de valgmuligheter de står overfor. Dessuten er det ønskelig å øke effektiviteten av helseinnsatsen ved å kanalisere tilbudet til dem med høyest kreftrisiko. I dag brukes det antakelig en betydelig mengde ressurser på kontroller av personer med lav risiko (vill-screening) fordi de ikke har fått en adekvat genetisk utredning.
Kontinuerlig evaluering av kontrollopplegget bidrar til kvalitetssikring og utvikling av et bedre tilbud. Derfor er det også en målsetting at helsevesenets data skal brukes til evaluering av virksomheten med adekvat metodikk.
Figur 3.3 viser en skjematisk framstilling av hvordan tilbudet til personer som tilhører familier med opphoping av kreft, er organisert i Norge i dag. Tilbudet består i hovedsak av følgende:
genetisk utredning (omtalt i punktene 3.3.1 og 3.3.2);
genetisk veiledning (omtalt i punkt 3.3.3);
tilbud om oppfølging (omtalt i punktene 3.3.4 og 3.3.5); og
gentesting for enkelte kreftformer (omtalt i kapittel 6).
Disse tilbudene gis i samarbeid mellom flere sykehusavdelinger i ulike helseregioner. Koordineringen av tilbudet styres som regel fra regionens medisinsk-genetiske enhet.
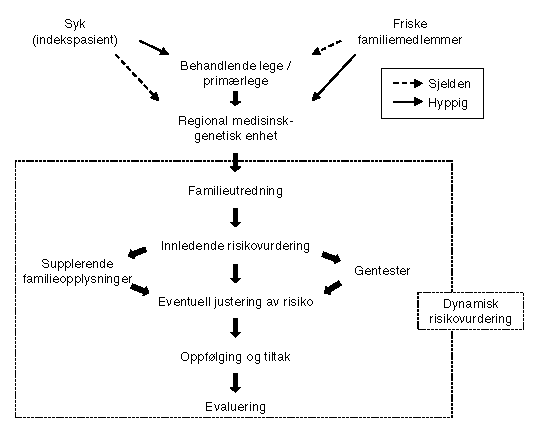
Figur 3.3 Flytskjema for utredningen av personer med antatt høy risiko for å utvikle kreft.
3.3.1 Genetisk utredning
Genetisk utredning omfatter innsamling av relevant informasjon (herunder familieopplysninger), diagnostiske overveielser og risikovurdering.
De fleste som hittil har henvendt seg til medisinsk-genetiske enheter, er friske familiemedlemmer som skriver direkte til enhetene med spørsmål om familiens opphoping av kreft. De har gjerne hørt at et slikt tilbud eksisterer fra slektninger eller andre. I den siste tida har også henvendelser fra primærhelsetjenesten og sykehusleger økt.
Det er altså i hovedsak to måter risikopersoner kommer i kontakt med medisinsk-genetiske enheter på: enten ved at de selv tar kontakt, eller ved at lege henviser dem (se figur 3.3).
3.3.1.1 Hvordan få pålitelige opplysninger om familiens sykdommer?
De første opplysningene om familiens diagnoser kommer fra indekspersonen (det familiemedlemmet som først tar kontakt med en medisinsk-genetisk enhet). I medisinsk-genetiske enheter innhentes opplysningene ved hjelp av standardiserte slektsskjema.
Grunnlaget for en riktig vurdering av risiko for arvelig sykdom, er korrekte diagnoser. For å kunne gjøre en tilfredsstillende genetisk utredning av familien, bør som hovedregel alle kreftdiagnoser i familien verifiseres. Det finnes ulike instanser hvor dette kan gjøres. Førstevalget er det sykehuset hvor den aktuelle pasient ble behandlet, eventuelt diagnostisert.
Legen har taushetsplikt i følge legeloven og kan følgelig ikke gi opplysninger om en pasient til familiemedlemmer (se også punkt 4.2.5.2). Det finnes likevel unntaksbestemmelser, først og fremst når pasienten selv samtykker. Samtykket må være informert. Det innebærer at pasienten forut for samtykket må ha forstått hvilke opplysninger det er snakk om, hva hensikten med opplysningene er, og konsekvensene av å oppheve legens taushetsplikt. Dersom det affiserte familiemedlemmet er i live, innhentes skriftlig samtykke til innsyn i hans eller hennes sykehusjournal. Samtykket gjelder kun de aktuelle kreftdiagnoser.
Dersom den affiserte er død, veier ikke personvernhensyn like tungt. Kravet til begrunnelse for å meddele taushetsbelagt helseopplysning er da redusert til vektige grunner, jf. legeloven § 37. Legen skal ta hensyn til arten av de opplysninger det dreier seg om, avdødes formodede vilje og de pårørendes og samfunnets interesser. Etter en konkret vurdering vil bestemmelsen kunne gi adgang til å gi opplysninger til den avdødes slektninger. Likevel hjemler heller ikke denne bestemmelsen for noen systematisk virksomhet; legen må etter konkrete vurderinger ta stilling til hvorvidt legeloven gir adgang til å gi slik informasjon. Praksis er at man kun verifiserer de kreftdiagnoser som, etter en totalvurdering av familien, gir mistanke om en arvelig komponent.
Dersom det ikke lar seg gjøre å verifisere de aktuelle diagnosene via sykehusjournalene, kan man forsøke hos Kreftregisteret. Kreftregisteret har ikke etablerte servicefunksjoner overfor helsevesenet. Dette medfører at henvendelser for å verifisere diagnoser må skje via personen som selv har kreftdiagnosen. Dersom vedkommende er død, kan opplysninger gis direkte til medisinsk-genetisk enhet.
Ikke alle diagnoser lar seg verifisere via Kreftregisteret. Det kan skyldes at diagnosen ble stilt før registeret startet sin registrering i 1952, eller, i sjeldne tilfeller, at kreftdiagnosen ved en forglemmelse ikke ble innrapportert. Det eksisterer i så fall et tredje alternativ for verifisering av diagnoser, nemlig via avdelinger for patologi. Reglene for personvern og skriftlig samtykke gjelder også her. De patologiske avdelingene har en betydelig samling av biopsier, blant annet fra tumorvev, som i enkelte tilfeller er over 100 år gamle. Dersom det foreligger usikkerhet om en diagnose som er stilt for mange år siden (for eksempel i forhold til klassifisering av svulst eller sykdom), kan patologene i noen tilfeller se på en gammel vevsprøve i lys av ny kunnskap og teknikk. Dette har i enkelte tilfeller ført til en revurdering av hele familiers risiko for å utvikle kreft.
3.3.1.2 Prioritering til kontrollopplegg
For de hyppigste kreftformene er det utarbeidet kriterier som kan bidra til å gi mistanke om arvelig kreft. Dersom familien tilfredsstiller kriteriene, eller den medisinsk-genetisk enheten finner grunn til det utfra en totalvurdering, får risikopersoner tilbud om kontrollopplegg (omtalt i 3.3.4) Kriteriene følger internasjonalt anerkjente kjennetegn på familier der det er sannsynlig at kreftsykdom går i arv.
De fleste av de kriteriene som siteres nedenfor, ble opprinnelig lansert i forskningsøyemed (for å skaffe til veie familier der det fantes et minimum av informasjon med tanke på såkalte koplingsanalyser). Kriteriene bygger i hovedsak på at sannsynligheten for at det dreier seg om arvelige tilstander er større jo flere familiemedlemmer som er affiserte, og jo yngre disse er. De ulike kriteriene vil kunne justeres noe som følge av nye erfaringer og viten.
I de fleste helseregioner i Norge vil en person som tilhører en familie med opphoping av kreft, få tilbud om genetisk veiledning selv om familien faller utenfor slektskriteriene. Men når det er spørsmål om hvem som skal inkluderes i kontrollopplegg, holder man seg som regel til kriteriene. Den genetisk utredningen innebærer likevel en individuell vurdering av alle pasienter og risikopersoner. Kriterier for mistanke om arvelig kreft er en rettesnor, men det blir alltid utført en totalvurdering av hele familien og de enkelte medlemmene.
Bryst- og eggstokkreft
Det som karakteriserer familier med arvelig brystkreft, er at mange av kvinnene får sykdommen, og at de er unge når sykdommen debuterer – gjerne i 30-40 års alderen. I noen av familiene er risikoen også stor for eggstokkreft. Det er utarbeidet nasjonale kriterier for mistanke om arvelig bryst- og eggstokkreft. 2 Kriteriene stemmer godt overens med praksis i andre land.
Kvinner med følgende familiehistorie bør henvises til medisinsk-genetiske enheter:
brystkreft før 45 år hos førstegradsslektning;
brystkreft før 55 år hos to førstegradsslektninger;
fire nære slektninger med brystkreft;
bilateral (tosidig) brystkreft før 60 år;
brystkreft hos førstegradsslektninger før 60 år dersom personen selv, eller annen slektning, har eggstokkreft;
flere nære slektninger med eggstokkreft; eller
påvist mutasjon i bryst- og/eller eggstokkreft-gener hos personen selv eller hans eller hennes foreldre.
Familiær ikke-polypøs kolorektal cancer (HNPCC)
Diagnostisering av HNPCC er ikke like enkelt som en del andre former for tykk- og endetarmkreft, for eksempel familiær adenomatøs polypose (FAP). En fullstendig og detaljert medisinsk familiehistorie, som inkluderer aldre ved sykdomsdebuter i familien, er et meget nyttig redskap for å identifisere risikogrupper som bør tilbys kontrollopplegg. Ei internasjonal arbeidsgruppe for HNPCC ble etablert i 1989. Den har sin administrative base nært knyttet opp mot The Netherlands Foundation of the detection of hereditary tumores i Leiden. En av oppgavene denne gruppa har, er å utarbeide retningslinjer for ivaretakelse av risikopersoner fra HNPCC-familier. Gruppa har også utarbeidet kriterier for HNPCC-diagnosen, de såkalte Amsterdam-kriteriene:
til sammen minst tre syke personer med tykk- eller endetarmkreft, hvorav minst én må være førstegradsslektning av de to andre, i minst to generasjoner, og minst en av dem må ha vært under 50 år ved sykdomsdebut; og
familiær adenomatøs polypose (FAP) må være utelukket.
Det er en svakhet ved disse kriteriene at de verken tar hensyn til familiestørrelse eller andre HNPCC-relaterte kreftformer. Kriteriene er derfor utviklet videre, og i Norge følger man i hovedsak de reviderte Amsterdam-kriteriene. I følge disse skal kreft i livmorens slimhinne telle like mye som tykk- og endetarmkreft ved beregning av risiko for HNPCC.
Mange tilfeller av arvelig tykk- og endetarmkreft vil falle utenfor kriteriene, simpelthen fordi familiene er for små. Kravet om minst tre syke vil dermed ikke være innfridd. Dette understreker betydningen av at man i den genetiske utredningen gjør en totalvurdering av familien.
De fleste familiene med opphoping av tykk- og endetarmkreft, innfrir ikke kravene til HNPCC. I disse familiene vil ofte krefttilfellene debutere senere i livet enn i HNPCC-familiene. Slike familier har fått betegnelsen sent debuterende CRC (tykk- og endetarmkreft med sent utbrudd).
Familiær adenomatøs polypose (FAP)
Det finnes et nasjonalt register for denne formen for tykk- og endetarmkreft, nemlig Norsk Polyposeregister (omtalt under punkt 3.4.3.5). Polyposeregisteret er i dag underlagt Kreftregisteret. Tilsvarende registre er etablert i en rekke europeiske land.
Bakgrunnsrisikoen for å utvikle polypose er meget lav i Norge. Utenlandske undersøkelser viser en populasjonsfrekvens på 0,3%. Man regner i Norge med én nyoppstått mutasjon hvert år (hos personer uten familiehistorie med polypose).
Forekomsten av én førstegradsslektning med polypose, uansett alder ved sykdomsdebut, er tilstrekkelig for å bli inkludert i kontrollopplegg for risikopersoner.
Prostatakreft
Mindre enn 5% av all prostatakreft antas å være arvelig. I familiær prostatakreft er det oftest et fåtall (1-2) syke, og tidlig sykdomsdebut (under 50 år) forekommer sjeldent. Få pasienter ses av de medisinsk-genetiske enhetene per i dag. Tilstanden oppfattes som sannsynlig arvelig dersom
to førstegradsslektninger (brødre eller far/ sønn) har prostatakreft før 65 års alder;
det er minst tre syke i familien; og
noen av de syke har debutalder før 55 år.
3.3.2 Bestemmelse av risiko for friske familiemedlemmer
3.3.2.1 Risikovurdering generelt
Risikovurdering betyr i denne sammenheng å finne sannsynligheten for at friske familiemedlemmer utvikler sykdom (eller at syke utvikler kreft i nye organer). Risikovurderingen danner grunnlaget for hvorvidt en person skal få tilbud om oppfølging og kontroller.
Alle metoder for risikovurdering har sine svakheter, og det er nødvendig å være innforstått med svakhetene for å unngå betydelige feil. Dette gjelder også dataprogrammer. Alle metodene krever derfor spesiell bakgrunn og erfaring hos dem som skal foreta beregningen. Vanligvis er de spesialister i medisinsk genetikk.
Generelt ved arvelig kreft vurderes risiko for friske ut fra en komplett og dokumentert slektstavle. Fordelingen av syke i familien gir holdepunkt for hvilket arvemønster som er sannsynlig. Familier med mange syke, eller med opphoping av unge syke, gir holdepunkt for dominant arv, og det er også den vanligste arveform ved kreft. Vurderingen skjer på grunnlag av generell genetisk kunnskap. Spesielle problemer oppstår i små familier eller i familier der sykdommen har hoppet over en generasjon (noe som av og til forventes, dersom genet har nedsatt penetranse).
Dersom dominant arv legges til grunn, har friske førstegradsslektninger (barn og søsken) av pasienter med påvist mutasjon 50% risiko for selv å være genbærer. Denne risikoen korrigeres for redusert penetranse og for den del av risikoperioden som den friske risikoperson allerede har gjennomlevd. Følgende kan tjene som et forenklet eksempel: 50% risiko for å være genbærer gir, etter korreksjon for penetranse, 40% risiko for sykdom for en 30-årig kvinne fram til alder 70 år. Dersom kvinnen er 50 år, har hun en risiko på ca. 20% fram til 70 år, siden hun grovt sett har levet gjennom halve risikoperioden. Til grunn for en slik utregning ligger betingede sannsynligheter.
Dersom det ikke kan legges til grunn et dominant arvemønster for familien, må empiriske risikotall danne grunnlaget for risikovurderingen. Dette innebærer å se på hva som faktisk har skjedd i familier med ulike konstellasjoner av kreft i publiserte kliniske materialer. Denne metoden har alltid svakheter og må brukes med betydelig varsomhet. Ofte finnes ikke empiriske tall for den aktuelle familiekonstellasjon, og man må intrapolere eller ekstrapolere.
Det er dessuten alltid en betydelig usikkerhet forbundet med bruken av tall fra populasjoner som ikke er norske. Genfrekvenser varierer sterkt populasjoner imellom, og man vet at den norske befolkningen avviker sterkt fra andre når det gjelder hyppigheten av BRCA1-mutasjoner. Også innen vårt land er det store forskjeller, så tall fra Oslo bør ikke brukes for Rogaland.
Tabeller basert på empiriske risikotall har også ulike svake sider fordi flere typer relevant informasjon er utelatt (for eksempel risikopersonens alder, alderen til de syke, at ikke alle syke er inkludert eller at antall friske ikke er tatt med). Dette understreker betydningen av at en god risikovurdering bare kan gjøres av en person med den nødvendige innsikt.
3.3.2.2 Risikovurderinger spesielt
Bryst- og eggstokkreft
De fleste risikotabeller for familier uten kjent mutasjon, angir kun risiko for brystkreft. Claus? tabeller, som er mye brukt, gir for eksempel bare risiko for brystkreft. Shattuck-Eidens? tabeller gir risiko for at en syk er BRCA1-bærer og omfatter også eggstokkreft. Vanligvis beregnes risiko til alder 70 år. Bakgrunnsrisiko i befolkningen (9% for brystkreft) er som regel lagt inn i en samlet risiko. Det antas at rene eggstokkreft-familier er sjeldne, og at det er tilfeldigheter som gjør at enkelte familier bare har syke med eggstokkreft. Selv i slike familier er risikoen for brystkreft større enn risikoen for eggstokkreft.
Påvisning av familiens mutasjon hos de syke er den sikreste metoden til å avgjøre hvorvidt kreft går i arv i en familie. Dette er en viktig grunn til å finne mutasjoner. Friske førstegradsslektninger av syke med påvist BRCA1-mutasjon (søsken og barn) har 50% sannsynlighet for å være genbærere, andregradsslektninger 25%. Risiko for sykdom er dette tallet multiplisert med mutasjonens penetranse, som er ca. 80% for brystkreft og ca. 60% for eggstokkreft (tallet varierer mellom materialer). Det må korrigeres for alder på risikopersonen (det vil si gjennomlevd del av risikoperiode).
Penetransen antas å variere fra mutasjon til mutasjon, men det vil ta tid før de forskjellige mutasjonenes penetranse er empirisk bestemt. For sjeldne mutasjoner, særlig de som bare finnes i én familie, vil det ofte være umulig å angi empiriske tall for penetranse.
Ved negativ test hos en frisk person i en familie med kjent mutasjon, er risikoen trolig nær befolkningens bakgrunnsrisiko. I slike tilfeller er det rimelig å avslutte kontrollopplegg.
Familiær ikke-polypøs kolorektal cancer (HNPCC)
I familier uten påvist mutasjon, men som fyller Amsterdam-kriteriene (beskrevet i 3.3.1.2), legges det til grunn at friske førstegradsslektninger har 50% risiko for å være genbærere, korrigert for ca. 80% penetranse, og eventuelt også for risikopersonens alder. Samlegruppa late onset CRC (sent debuterende kolorektal cancer) er nylig avgrenset, og det er derfor vanskelig å angi risiko. Gode, empiriske tall for risiko mangler.
Wijnen gir tabeller for risiko for HNPCC. 3
For friske med påvist mutasjon i MLH1/ MSH2-genene, er sykdomsrisiko ca. 80%.
3.3.2.3 Risiko som tilsier tiltak
Grenseverdien for risiko som skal følges opp av forebyggende tiltak (se punkt 3.3.4), er satt til 25% livstidsrisiko for å bli syk (2-3 ganger bakgrunnsrisikoen for brystkreft).
Ved formidling av risiko til familiene brukes vanligvis et beskrivende uttrykk i stedet for tall: bakgrunnsrisiko, moderat forhøyet risiko og høy risiko.
3.3.2.4 Risikovurderingens dynamikk
Et risikotall gjelder bare med de premisser som ble lagt til grunn for det. Når premissene endres, så endres også risikoen. Den blir således redusert med økende alder fordi en økende del av den biologiske risikoperioden er tilbakelagt. For arvelig brystkreft er risikoen omtrent halvert fra 30 til 50 års alder. Men dersom det oppstår nye krefttilfeller i familien, vil risikoen kunne øke sterkt.
I tillegg endres ofte den antatte penetransen for genet ettersom bedre populasjonsdata legges fram. Dette har skjedd for BRCA1; etter at de første tallene ble presentert, har andre pasientgrupper gitt forskjellige penetransedata. For de hyppige mutasjonene vil det etter hvert komme mutasjonsspesifikke penetranseverdier som kan være andre enn genets samlede penetranse. Risikovurderinger endres altså over tid og må mange ganger revurderes.
3.3.3 Genetisk veiledning
3.3.3.1 Definisjon av genetisk veiledning 4
Genetisk veiledning er en kommunikasjonsprosess som tar for seg menneskelige problemer forbundet med forekomst, eller risiko for forekomst, av arvelig sykdom i en familie. Denne prosessen omfatter forsøk av en eller flere spesielt utdannede personer på å hjelpe individet og/eller familien til:
å forstå de medisinske fakta, inklusive diagnosen, den sannsynlige utvikling av sykdommen, og de tilgjengelige behandlingsmuligheter;
å forstå hvordan arvelige faktorer bidrar til forekomst av sykdommen/tilstanden, og hvordan man utfra dette kan fastslå risiko for gjentakelse for ulike slektninger;
å forstå valgmulighetene som finnes for å leve med eller omgå den risiko for gjentakelse som beskrives;
å velge den handlemåte som synes adekvat i lys av den enkeltes risiko, familiemålsetting, etiske og religiøse overbevisninger, og derved støtte familien i beslutninger; og
å tilpasse seg sykdommen hos familiemedlemmet og risikoen for at sykdommen skal kunne opptre hos barn eller andre familiemedlemmer.
3.3.3.2 Målsetting for genetisk veiledning
Målsettingen med genetisk veiledning er å sette familien og den enkelte i stand til å forstå sine helseproblemer, slik at de kan fatte beslutninger på et best mulig informert grunnlag. For å oppnå dette, er man avhengig av et grundig utredningsarbeid. I utredningsarbeidet er blant annet den medisinsk-genetiske kunnskapen fra en spesialist i medisinsk genetikk helt avgjørende. Takst for genetisk veiledning avspeiler også at veiledningssamtalen er uløselig knyttet til utredningen. I samtalen formidles resultatet fra den genetiske utredning til den aktuelle personen. Veilederen tilstreber i løpet av en genetisk veiledningssamtale å være mest mulig nøytral vis-à-vis personens valgmuligheter.
Sentralt i den genetiske veiledning ved arvelig kreft, står opplysninger om effekten av tiltakene. Hensikten med tiltakene er å forebygge, oppdage og behandle kreftsykdommer.
Definisjonen ovenfor legger stor vekt på prosessen. Denne er tidkrevende, og kan vanskelig oppfylles i løpet av én konsultasjon eller av én person alene. For å kunne treffe beslutninger, må pasienten ha tilstrekkelig informasjon. Informasjonen må være tilpasset mottaker, så veiledningen foregår som regel i et 1:1 forhold (én veileder og én pasient). Det er likevel ikke uvanlig at familiemedlemmer ønsker å komme sammen til veiledningssamtaler. Selvetablerte grupper kan ha sine fordeler, for eksempel i form av innbyrdes kommunikasjon i etterkant av samtalen. I forbindelse med gruppeveiledninger, blir som regel medlemmene av gruppa også tilbudt en kortere individuell veiledning.
Veiledning er nødvendig blant annet for at risikopersoner eller pasienter skal kunne gi et informert samtykke, for eksempel i forhold til framtidige gentester. Dagens lovverk krever at det skal gis omfattende genetisk veiledning i forbindelse med undersøkelser for arvelig sykdom (se punkt 4.2.2.5). Den som skal gi slik veiledning, må ha spesiell kompetanse innen medisinsk genetikk og god innsikt i problemstillingene. I tillegg må sykdommen og familieanamnesen på forhånd være grundig utredet, og veileder må ha konsultert spesiallitteratur og vurdert eventuell supplerende klinisk og laboratoriemessig diagnostikk.
Den vanligste grunnen til at folk ønsker genetisk veiledning, er at de eller noen av deres nærmeste er rammet av noe de vet eller frykter er arvelig sykdom. De fleste som selv søker genetisk veiledning, ønsker svar på spørsmål om hva som har hendt og hvorfor; om det vil kunne hende igjen; og hva de kan gjøre for å modifisere risikoen.
3.3.4 Tilbud om kontrollopplegg
Etter genetisk utredning og genetisk veiledning, vil aktuelle personer få tilbud om kontrollopplegg. Hensikten med kontrollopplegget er å oppdage sykdommen på et forstadium eller før den har spredd seg. Sjansen for helbredelse er betydelig redusert ved spredning.
Prosessen fra en person tar kontakt med en medisinsk-genetisk enhet, og til et eventuelt kontrollopplegg er klart, er tidkrevende. Utredning, blant annet med verifisering av familiens ulike kreftdiagnoser, tar tid. Det vurderes også fortløpende om det vil være nødvendig eller ønskelig med en eller flere oppfølgingssamtaler etter en genetisk veiledningssamtale. Slike tilbud er både tid- og ressurskrevende.
Inntil for få år siden var det i hovedsak sjeldne tilstander som ble ivaretatt i medisinsk-genetiske enheter. Med den nye genteknologien har store folkesykdommer (som kreft og hjerte- og karsykdommer) blitt aktuelle, og disse utgjør en stadig økende del av den medisinsk-genetiske virksomheten. I det følgende omtales tilbudet for de mest utbredte arvelige kreftformene.
3.3.4.1 Tilbud til familier med opphoping av bryst- og/eller eggstokk kreft
De følgende retningslinjene er utarbeidet av Norsk Bryst Cancer Gruppe (NBCG), 5 og er i samsvar med retningslinjer man følger ellers i Europa. Unge kvinner med vesentlig økt risiko for å utvikle brystkreft, bør undersøkes på poliklinikker med adekvat medisinsk kompetanse og tilstrekkelige diagnostiske muligheter. Kvinner som faller inn under gjeldende kriterier vil, om de ønsker det, inkluderes i et slikt kontrollopplegg.
Den anbefalte oppfølging av kvinner med antatt høy risiko for å utvikle brystkreft, er en årlig klinisk undersøkelse av brystene hos lege med spesialkompetanse i for eksempel brystkirurgi/ gynekologi. Legen må samarbeide med en erfaren røntgenlege som foretar mammografi, og det skal gjøres tilleggsundersøkelser på liberal indikasjon (for eksempel ultralyd av brystene og/eller vevsprøver). Undersøkelsene starter ved 30 års alder. Det anbefales for tida årlige undersøkelser inntil kvinnen er 55 år. Deretter overføres hun til mammografi-screening (mammografi-undersøkelser annethvert år til fylte 69 år). Postmenopausale, predisponerte kvinner har fortsatt en høyere kreftrisiko enn jevnaldrende kvinner, så supplerende diagnostiske tiltak bør også her utføres på liberale indikasjoner.
Ved samtidig risiko for eggstokkreft i familien, samordnes kontrollopplegget med gynekologisk undersøkelse. De gynekologiske kontrollene består av årlig vaginal ultralyd og måling av CA 125 (en biomarkør som finnes i blodet hos mange kvinner med eggstokkreft) fra kvinnene er 35 år. Dette kontrollopplegget varer i prinsippet livet ut. I visse tilfeller bør en vurdere kirurgisk fjerning av eggstokkene ved 40-45 års alder. Undersøkelsene rekvireres etter bosted, ved nærmeste sykehus med tilstrekkelige ressurser.
Effekten av brystkontrollene er delvis dokumentert; og alt tyder på at oppfølgingsopplegget er i stand til å oppdage brystkreft på et meget tidlig stadium. Ett forskningsprosjekt viste at kontrollopplegget fant i underkant av ett prospektivt brystkrefttilfelle per 100 undersøkte kvinne. 6 Prosjektet viste også at 87% av krefttilfellene ble oppdaget uten spredning til lymfeknute. Endringen av dødeligheten er imidlertid foreløpig ukjent, men hovedregelen har hittil vært at jo tidligere en oppdager en kreftsvulst i brystet, jo bedre er prognosen. 7
Nytten av eggstokk-kontrollene er foreløpig ukjent.
Oppsummering av kontrollopplegg for arvelig bryst- og eggstokkreft:
årlige kliniske brystundersøkelser og mammografi-undersøkelser fra kvinnen er 30 år til 55 år; og
ved risiko for eggstokkreft: årlig gynekologisk undersøkelse med vaginal ultralyd og måling av CA125 fra kvinnen er 35 år og i prinsippet livet ut.
3.3.4.2 Tilbud til personer med forhøyet risiko for kreft i tykk- og endetarm
Kontroll for arvelig non-polypøs tykktarmkreft (HNPCC)
Anbefalt kontrollopplegg for risikopersoner som tilhører HNPCC-familier i Norge er:
koloskopi hvert annet år fra 30 års alder, eller 5 år før tidligste debut i familien; og
for kvinner er det i tillegg kontroll av endometriet (livmorens slimhinne), med vaginal ultralyd og måling av CA125, hvert annet år fra 35 års alder. 8
Effekten av kontrollene er delvis dokumentert. 9
Norsk gynekologisk forening har utarbeidet retningslinjer for kontroll av endometriet hos kvinner som tilhører HNPCC-familier. 10 Både koloskopi og endometriekontroller tilbys livet ut. Tilsvarende kontrollopplegg er satt i verk i flere europeiske land og i USA.
For familier med opphoping av tykk- og endetarmkreft som ikke dekker kravene til HNPCC (sent debuterende CRC), er kontrollopplegg ikke avklart. Det er imidlertid foreslått koloskopi hvert 5. år fra risikopersonene er 40 år. Hver familie må i større grad enn ellers vurderes individuelt.
Kontrollopplegg for familiær adenomatøs polypose (FAP)
Familiemedlemmer med risiko for å utvikle polypose tilbys
tarmundersøkelser (endoskopi).
Polyppene kan oppdages i tykk- og endetarmen på et tidlig stadium, og behandlingen settes inn før det utvikler seg kreft. Kontroll av endetarmen og nedre del av tykktarmen starter ved 10-års alderen. Så lenge det ikke er påvist polypper, anbefales det å fortsette disse kontrollene annet hvert år til risikopersonen er 40 år. Fra 41 til 50 års alder anbefales det kontroller hvert tredje år, og deretter hvert femte år fram til 60-års alderen. Først da kan en være rimelig sikker på at sykdommen ikke vil utvikle seg. Både øvre og nedre endoskopi-undersøkelser kan bli benyttet ved disse kontrollene.
Hos barn som er disponert for polypose kan det finnes små mørke pigmentflekker på netthinnen som gir et forvarsel om utviklingen av sykdommen. Erfarne øyespesialister vil kunne oppdage disse forandringene, men barna må være noen år før det gjennomføres
undersøkelse av øyebunnen (oftalmoskopi).
3.3.4.3 Tilbud til personer med forhøyet risiko for kreft i prostata
Det finnes ikke noe felles opplegg for tiltak for denne gruppa. Personer som faller inn under kriteriene for sannsynlig arvelig tilstand (se punkt 3.3.2.1), får tilbud om årlig undersøkelse hos urolog med eksplorasjon, transrektal ultralyd og måling av PSA (en kreftindikator som kan måles i en blodprøve). Tiltaket starter ved 55 års alder, eller 10 år før yngste sykdomsdebut i familien. Familiene følges med tanke på eventuell forskningsmessig bearbeidelse for å skaffe kunnskap om tilstanden.
3.3.5 Forebyggende tiltak
En mulighet for personer med høy risiko for kreft, er å fjerne risiko-organer profylaktisk (forebyggende), etter en avveining av risikoen for å dø av sykdommen mot graden av lemlestelse og andre bivirkninger av inngrepet. Profylaktisk kirurgi er etablert praksis for familiær tykktarmpolypose; her fjernes tykktarmen i ung alder fordi risiko for kreft ubehandlet er nær 100%. Det samme gjelder for familiær skjoldbruskkjertelkreft.
Ved høy risiko for eggstokkreft anbefales det å fjerne eggstokker fra 45 års alder, men kvinnen og gynekologen tar avgjørelsen etter en totalvurdering. Det er ikke dokumentert at kontrollopplegget påviser eggstokkreft tidlig nok for helbredende kirurgisk behandling.
Det finnes derimot utstrakt tilbakeholdenhet med å fjerne bryster forebyggende. Inngrepet oppfattes som dramatisk, og kontrollopplegget for brystkreft er rimelig godt (ca. 90% av tilfellene blir funnet i tide). Men det finnes holdepunkter for at visse BRCA1-mutasjoner kan gi en kreftform med spredning ved diagnosetidspunktet og derfor dårlig prognose. I slike sammenhenger vurderes profylaktisk fjerning av bryster som et reelt alternativ. I visse land (Frankrike) har holdningene til profylaktisk kirurgi allerede endret seg.
Det drives omfattende forskning for å finne medikamenter som forebygger brystkreft hos kvinner med høy risiko. Tamoxifen har vært prøvd, men forholdet mellom effekt og bivirkninger har vist seg for dårlig. Andre medikamenter utprøves, men per i dag er ikke noe anbefalt medikament registrert.
3.3.6 Genetisk testing av barn
Genetisk testing av barn er omtalt i lov om medisinsk bruk av bioteknologi (se punkt 4.2.2.5). Loven deler testingen inn i a) diagnostisk testing, b) presymptomatisk testing, c) prediktiv testing og d) arvebærertesting. Anbefalingene som er beskrevet under er hentet fra en rapport fra Clinical Genetics Society (Storbritannia) 11 . De er i overensstemmelse med lov om medisinsk bruk av bioteknologi. Det forutsettes hele tida at barnets foreldre samtykker.
3.3.6.1 Diagnostisk testing
Dersom barnet allerede har tegn på familiær sykdom eller tilstand med kjent kreftrisiko, er det ansett som uproblematisk å utføre gentest såframt det dreier seg om tilstander der det regelmessig opptrer sykdomssymptomer og -tegn i barnealder. Dersom det finnes virksom behandling (som diett, medikamenter eller muligheter for å hindre komplikasjoner) er det ansett som indisert å teste barn med foreldrenes samtykke.
3.3.6.2 Presymptomatisk og prediktiv testing
Dersom det derimot dreier seg om en tilstand med start i voksen alder og manglende behandlingsmulighet, skal testing ikke skje før barnet selv anmoder om det og er myndig, eventuelt over 16 år. Grunnen til dette er at respekten for konfidensialitet og autonomi tilsier at man venter til individet kan gi et informert samtykke. Dette krever innsikt ikke bare i de genetiske, men også de følelsesmessige og sosiale implikasjoner av testing og testresultater. Det betyr ikke at testens ulike implikasjoner – inkludert eventuell testing av andre familiemedlemmer – ikke bør drøftes før voksen alder, men at selve testingen bør vente til da.
Det finnes tilstander der det er uklart om presymptomatisk testing er av betydning for barn som hittil har vært friske. Slik testing må i utgangspunktet være gjenstand for videre forskning, spesielt når det gjelder psykologiske og sosiale følger.
3.3.6.3 Arvebærertesting
Testing for bærerstatus for recessive tilstander og kromosomtranslokasjoner er mer komplekst. Bærertilstand for recessive sykdommer er bare av potensiell reproduktiv betydning. Testing for kromosomtranslokasjoner uten symptomer hos barn, kan teoretisk gi informasjon om foreldrenes tilstand og være avklarende for andre, om enn meget sjelden. Dersom testing ikke skjer i barnealder må det påhvile familien og helsevesenet en felles forpliktelse til å opplyse om testtilbud når barnet blir voksent. For mange sykdommer er det kanskje slik at en gentest vil være mindre nøyaktig og gi mindre informasjon nå enn om noen år, slik at oppdatering av test og genetisk veiledning kan være aktuelt senere.
Arvebærertesting av barn er lite aktuelt i forbindelse med kreftsykdommer.
3.4 Organisasjon av tilbudet til familier med risiko for arvelig kreft
3.4.1 Helsevesenets organisasjon
Det norske helsevesenet er inndelt i fem helseregioner (Øst, Sør, Vest, Midt-Norge og Nord) og flere nivåer (for eksempel lokalsykehus, sentralsykehus og regionsykehus). Den regionale og fylkeskommunale planleggingen legger til rette for en oppgavefordeling mellom sykehusene som tar hensyn til tjenestenes kvalitetskrav. Kvaliteten på helsetjenestene er derfor både et faglig og et organisatorisk anliggende. Medisinsk-genetiske tjenester, inkludert kreftgenetikk, skal i første rekke bygges opp som regionale funksjoner.
Ved inngangen til 1999 fantes det tre generelle, regionale medisinsk-genetiske enheter med alle servicefunksjoner (klinisk genetikk, DNA-laboratorium og cytogenetisk laboratorium) i Norge: Avdeling for medisinsk genetikk ved Ullevål sykehus, Avdeling for medisinsk genetikk ved Haukeland sykehus og Medisinsk genetisk avdeling ved Regionsykehuset i Tromsø. Det finnes også en avdeling for klinisk genetikk ved Rikshospitalet og en seksjon for medisinsk genetikk ved Radiumhospitalet. Radiumhospitalet har for tida ansvaret for kreftgenetikken i helseregionene Sør og Midt-Norge. I Nasjonal kreftplan er det foreslått brukt kr 100 millioner til opprettelse av kompetansesentre for arvelig kreft ved alle regionsykehus i perioden 1999-2003. 12
Tilbudet om genetisk utredning og veiledning gis fortrinnsvis ved nærmeste medisinsk-genetiske enhet. Oppfølging av risikopersoner (kontroller, se punkt 3.3.4) skjer ved nærmeste sykehus med tilstrekkelig kompetanse. Risikopersonene må i noen tilfeller forholde seg til svært mange aktører. Flere institusjoner på forskjellige nivåer kan være involvert i utredning, kontroller og behandling. Spesialisering innenfor de ulike sykehusene kan innebære en ekstra belastning, spesielt for personer med lang reisevei. En kvinne som er vurdert til å ha høy risiko for å utvikle bryst- og eggstokkreft, kan for eksempel risikere å bli tilbudt kontroller ved tre ulike seksjoner (røntgen, ultralyd-laboratorium og gynekologisk poliklinikk) på tre ulike dager. For mange vil dette innebære lange reiser og flere dager borte fra jobb og andre forpliktelser.
Dagens helsevesen ble i stor grad organisert på bakgrunn av kunnskapen om organpatologi. I prinsippet har hvert organ fått sin avdeling eller seksjon i sykehussystemet. Det er få instanser innen helsevesenet som er organisert rundt pasientens behov for ulike undersøkelser eller omsorgstiltak. Både i England og USA har man i større grad begynt å arrangere klinikker der leger fra ulike disipliner kommer sammen på gitte tider for å ta seg av ei pasientgruppe. Fordelen med slike klinikker er at pasientens ulike kontroller blir koordinert. Dessuten får de ulike disiplinene som ivaretar pasientgruppa et bedre grunnlag for samarbeid. Man kan dermed i større grad trekke veksler på hverandres erfaringer.
Samarbeidet mellom helseprofesjoner på tvers av forvaltningsnivåer og regionsgrenser, skaper ofte praktiske vansker for pasientene. Det er flere årsaker til dette. Medlemmer av en familie med opphoping av arvelig sykdom bor for eksempel ikke nødvendigvis innenfor grensen av en og samme helseregion. Trygdekontoret dekker i utgangspunktet utgifter til regionens medisinsk-genetiske enhet, og ikke til den enheten som har foretatt den primære vurderingen av familien. Det er ofte denne enheten som sitter inne med opplysninger både om familiens diagnoser, mutasjonsfunn i forbindelse med gentester, svar på eventuelle biokjemiske undersøkelser og liknende. Enkelte ganger kan familiers geografiske spredning føre til at flere genetiske enheter utreder samme familie uten å vite om det. Resultatet er unødig bruk av ressurser. Smidigere systemer og bedre samarbeid mellom de involverte enhetene vil kunne avhjelpe problemet, men faren for dobbeltregistrering av familier med opphoping av kreft er likevel betydelig.
Stadig flere behandlingsopplegg standardiseres. I praksis innebærer dette gjerne at man blir enige om nasjonale eller internasjonale retningslinjer for ivaretakelse av visse pasientgrupper. Tanken er at flest mulig skal følge disse behandlingsregimene eller kontrolloppleggene, blant annet fordi man da i neste omgang kan evaluere nytten. Slik er også pasientene sikret ensartede behandlingsopplegg uavhengig av bosted. Dette gjelder også for personer med høy risiko for å utvikle visse former for arvelig kreft som brystkreft, eggstokkreft og tykk- og endetarmkreft.
Mange av kontrolloppleggene som eksisterer i dag, er utilstrekkelig dokumentert. Den ønskede nytteeffekt av kontrolloppleggene, er reduksjon av dødeligheten gjennom tidlig oppdagelse og behandling. Vi vet en del om denne effekten ved mammografi-undersøkelser fra ulike norske undersøkelser. Det foreligger også resultater som viser at screening med koloskopi reduserer dødeligheten av tykk- og endetarmkreft blant risikopersoner i HNPCC-familier. Nytteeffekten av tarmkontroller hos risikopersoner for familiær adenomatøs polypose, har også vist seg å være gode. Effekten av eggstokk-kontroller er imidlertid ikke dokumentert.
Dersom kontrolloppleggene skal kunne evalueres, må de foregå innenfor rammen av prospektive undersøkelser. Det vil da være mulig å føre en kvalitetssikring av helsevesenets arbeid, både hva angår redusert dødelighet og psykososiale aspekter ved gentesting.
3.4.2 Journalpraksis
De medisinsk-genetiske enheter i Norge oppretter egen journal for alle personer som mottar genetisk veiledning. Disse journalene holdes atskilt fra sykehusets hovedjournal. Det er vanlig praksis at de genetiske enheter ikke låner ut journalene til andre enheter ved sykehuset. Når det gjelder håndteringen av arvelig kreft, blir det ved de fleste genetiske enheter også opprettet et familie-registreringsnummer dersom mer enn én person i en familie har henvendt seg til enheten. Journalene for hver familie arkiveres samlet, men det enkelte familiemedlem har bare innsynsrett i sin egen journal.
Alle journaler er arkivert i forskriftsmessige journalarkiv i de respektive medisinsk-genetiske enheter. Det finnes også elektroniske støttejournaler. Grunnen til den relativt strenge journalpraksisen, er at genetisk informasjon oppfattes som særlig sensitiv. Genetisk informasjon handler ikke bare om en persons sykdom, men også om hans eller hennes risiko for framtidig sykdom og sågar kommende generasjoners sykdomsrisiko. I frykt for at slik informasjon kan komme på avveier, praktiseres strenge retningslinjer.
3.4.3 Omsorg for belastede familier
3.4.3.1 Omsorgsbegrepet
Det er ikke alltid like klart hva som ligger i omsorgsbegrepet. Ofte benyttes det som en samlebetegnelse på alle medisinske funksjoner som er knyttet til ei diagnosegruppe, for eksempel kreftomsorgen: forebygging, diagnostikk, behandling, kontroller etc. Utvalget forstår imidlertid omsorgsbegrepet som en betegnelse på den form formidlingen av helsetjenester har, snarere enn som en egen helsetjeneste.
At en helsetjeneste formidles med omsorg innebærer blant annet at den formidles med med-menneskelighet, respekt, engasjement, innlevelse og omtanke.
Alle profesjoner og fagmiljøer innenfor helsevesenet har rettigheter og forpliktelser overfor omsorgsbegrepet. Formidling av omsorg krever blant annet erfaring, men også kunnskap som kan og må læres.
Omsorg, forstått som den form helsetjenester bør formidles på, må utvikles av helsepersonell i samråd med pasienter, pårørende og familie. God omsorg er sterkt knyttet til pasientens og de pårørendes opplevelser. Den kan og må læres, blant annet fordi kommunikasjon er et viktig element i formidlingen av god omsorg. Man er således på dette feltet avhengig både av forskning og av planmessig kunnskapsformidling til helsearbeidere.
Omsorg utgjør en kjerne i verdigrunnlaget og skal derfor være en felles basis for all behandling, pleie og kontakt mellom helsearbeidere, pasienter og pårørende.
3.4.3.2 Omsorg innenfor medisinsk genetikk
Medisinsk genetikk er en relativt ung disiplin innen medisinen og et fagfelt som ekspanderer meget raskt. Genteknologien står sentralt innen forskningen, også på folkesykdommer som kreft. Ny kunnskap fører til at stadig nye pasientgrupper vil søke genetisk veiledning og informasjon. Hvordan påvirker denne kunnskapen personer som søker genetisk veiledning for arvelig kreft? Påføres risikopersonene ytterligere bekymringer? Makter helsevesenet å ivareta personenes behov for informasjon og omsorg? Er tilbudet til risikopersoner med på å øke deres forståelse, innsikt og trygghet? Dette er spørsmål helsevesenet må forsøke å besvare.
I motsetning til folk flest, har alle risikopersoner opplevd alvorlig livstruende sykdom hos sine nære. Ved å tilby risikopersonene genetisk veiledning, vil kritikere hevde at man bygger opp under deres usikkerhet og angst og derved påfører dem ekstra bekymringer. Erfaringer fra genetiske enheter i Norge viser imidlertid at dette ikke er hovedregelen. Konsultandene angir stort sett at de er meget fornøyd med det tilbud de får. For mange av dem vil den genetiske veiledningssamtalen være første gang helsevesenet tar deres tanker og bekymringer rundt kreftsykdommen alvorlig. Mange av familiene med opphoping av kreft har kjent og erfart at kreft er arvelig, uten at de har fått gehør for dette i helsevesenet.
Forskningen hittil kan tyde på at individer som tilhører familier med opphoping av kreft allerede tenker mye på sin risiko, slik at genetisk veiledning snarere avdekker enn induserer nye bekymringer. Forskningsresultater viser også at det finnes personer med høy risiko for kreft som rapporterer et høyt angstnivå og lav livskvalitet, og som ofte overvurderer sin risiko. Mange får et mer avbalansert og realistisk forhold til sin risiko etter en genetisk veiledning. Det er imidlertid nylig publisert en britisk artikkel som tyder på det motsatte. 13 Dette forteller oss at man må være særdeles varsom, ydmyk og lydhør overfor brukernes behov for omsorg.
3.4.3.3 Hvordan imøtekommer helsevesenet risikopersonenes behov for omsorg?
Det er vanligvis de medisinsk-genetiske enhetene som er ansvarlige for å formidle informasjon om arvelige sykdommer og/eller gentester. I disse enhetene vil risikopersoner blant annet møte spesialister i medisinsk genetikk. De fleste medisinsk-genetiske enhetene i Norge har i tillegg tilsatt genetiske veiledere for å ivareta risikopersonenes omfattende behov for informasjon. Genetisk veileder er en egen yrkestittel i de fleste europeiske land og i USA ( genetic counsellor). Deres faglige bakgrunn er ulik, men mange av dem har en Master grad i genetic counselling. I Norge er ikke genetisk veileder en beskyttet tittel, og det foreligger ikke noen formelle krav til en slik stilling.
De medisinsk-genetiske miljøer i Norge har likevel klare forventninger til denne yrkesgruppa. For eksempel ønsker flere genetisk enheter at genetiske veiledere skal ha utdannelse på hovedfagsnivå innenfor et relevant fagfelt. De må også ha tilstrekkelig biologisk kunnskap og forståelse, så en helsefaglig bakgrunn er å foretrekke. Ved siden av den formelle utdannelsen, bør kandidaten ha flere års praktisk erfaring fra en medisinsk-genetisk enhet før vedkommende kan arbeide selvstendig. Disse kravene skyldes at genetiske veiledere har et stort ansvar (selvstendig genetisk utredning og veiledning under supervisjon av overlege). Det kreves betydelig formell kompetanse innen kommunikasjonsprosesser, pedagogikk og krisereaksjoner.
Genetiske veiledere har ikke som mål å bli eksperter i medisinsk genetikk, men å bli eksperter i å formidle genetisk kunnskap. Det eksisterer i dag ingen formell utdanning for genetiske veiledere i Norge, men det er startet innledende arbeid med en skandinavisk modell ved Nordiska Hälsovårdhögskolan i Göteborg.
Bioteknologiloven bidrar til å sikre brukere av genetiske tjenester visse rettigheter i forhold til informasjon. Dette gjelder både den omfattende genetiske veiledningen man har krav på i forbindelse med genetiske undersøkelser, og det informasjonsaspektet som ligger implisitt i det informerte samtykket. Prediktiv gentesting reiser en rekke viktige spørsmål av både etisk, juridisk og sosial art. For at risikopersonene skal kunne gi et informert samtykke, må de blant annet forstå at genetisk utredning, i likhet med mye annen informasjon i medisinen, i stor grad bygger på analyser av risiko (sannsynlighetsberegninger). Det er også viktig at de som skal testes er forberedt på hvilke reaksjoner de kan få når testresultatet formidles. Et positivt testresultat kan for eksempel innebære ugunstige psykologiske konsekvenser for dem selv og deres familie, samtidig som det gir dem en mer eksakt informasjon om deres risikoer.
Formidlingen av genetisk informasjon er en utfordring, ikke minst fordi genetikken i dag er relativt lite kjent blant folk flest. Omfattende veiledninger er derfor påkrevet, og enkelte ganger vil det også være behov for reveiledning. Dersom patologiske reaksjonsmønstre oppstår i forbindelse med genetisk veiledning eller oppfølging, må vedkommende henvises videre i helsevesenet på vanlig måte. Noen medisinsk-genetiske enheter har knyttet til seg psykiatrisk ekspertise i forbindelse med prediktiv gentesting.
Det er grunn til å tro at kreftbehandlingen, inkludert kreftomsorgen, vil endre seg i takt med ny kunnskap om kreftgenetikk. Det er således viktig å undersøke hvordan den nye kunnskapen påvirker de nye brukergruppene i helsevesenet, og hvilke psykososiale kostnader og gevinster slik kunnskap kan avstedkomme. Risikopersoner er ei potensielt sårbar gruppe som det er viktig å få kunnskap om, slik at helsevesenets intervensjoner kan planlegges best mulig. Klinisk forskning på denne pasientgruppa er nødvendig. Ved Radiumhospitalet pågår et omfattende forskningsprosjekt som søker å dokumentere livskvalitet ved bruk av gentester. Det er også meget viktig å få vurdert de sosialpolitiske sidene ved den genetiske revolusjon, blant annet individets rett til personvern og individuell selvbestemmelses- og eiendomsrett til genetisk informasjon.
3.4.3.4 Den Norske Kreftforening (DNK)
Av frivillige organisasjoner er det Den Norske Kreftforening som har størst betydning innen kreftomsorgen. DNK er en landsomfattende organisasjon med hovedkontor i Oslo og omsorgssentre i hvert fylke. DNK mottar ikke offentlig støtte; virksomheten er utelukkende basert på økonomisk støtte fra befolkningen. Til sammen har DNK mer enn 150.000 medlemmer. Foreningen har som mål å drive kreftforskning, informasjon og omsorg overfor pasienter og deres pårørende. Forskning er det største satsingsområde, og DNK er den største bidragsyter til kreftforskning i Norge.
Formålet med omsorgsvirksomheten er å være et supplement til det offentlige helse- og sosialtilbud. I tillegg ønsker DNK å bidra til bedret kvalitet på kreftomsorgen, og til en mer likeverdig fordeling av omsorgstilbudet til kreftpasienter i alle deler av landet. Andre viktige tiltak er samarbeidsprosjekter med den offentlig helsetjenesten og undervisning for offentlig helsepersonell. Foreningen driver dessuten en opplysningstelefon om kreft.
DNK utgir en rekke informasjonsbrosjyrer og tidsskrifter, noen med hele befolkningen som målgruppe, andre rettet mot spesielle grupper (pasienter, pårørende, helsepersonell, media etc.). Det produseres også brosjyrer som handler om hvordan kreftsykdom kan forebygges. I tillegg er det lagt vekt på faglige publikasjoner til helsepersonell og undervisningsmateriell til frivillige medhjelpere.
3.4.3.5 Norsk Polyposeregister
Norsk Polyposeregister ble opprettet som et nasjonalt register i 1978. Den Norske Kreftforening hadde det økonomiske ansvaret fram til 1994. Deretter har Sosial- og helsedepartementet, gjennom Kreftregisteret, overtatt ansvaret. Det er vanligvis regionsykehusene som melder personer til Polyposeregisteret.
Hensikten med Polyposeregisteret er å sørge for at alle pasienter og risikopersoner får et tilstrekkelig tilbud om omsorg og informasjon, rutineundersøkelser og eventuell behandling. Polyposeregisterets viktigste oppgave er således å koordinere kontroller for familier som er spredd over hele landet. Det sendes påminnelse om kontroll til behandlende leger, og opprettes kontakt mellom leger ved ulike sykehus dersom personer flytter. Kontrollene er forsøkt sentralisert til de enkelte regionsykehus. Geografiske og sosiale forhold kan i noen tilfeller vanskeliggjøre dette, og det gjøres individuelle vurderinger for å finne et passende kontrolltilbud til familiene.
De faglige retningslinjene er fastlagt og justeres i tråd med økende kunnskap om sykdommen. Ei referansegruppe av kirurger, indremedisinere og genetikere fra alle helseregionene har ansvaret for dette. Det er også opprettet et arbeidsutvalg. Registeret ønsker en sentral plass i nasjonal og internasjonal klinisk, genetisk og epidemiologisk forskning, for derved å øke kunnskapen om polyposesykdommen og forbedre behandlingen og oppfølgingen av de som blir rammet.
Medlemmer av polyposefamilier kan henvende seg direkte til Polyposeregisteret. Gjelder det medisinske spørsmål, vil sekretæren vise til rette instans. Videre veiledning vil da bli gitt i den regionen personen er hjemmehørende, for eksempel ved regionens medisinsk-genetiske enhet. Registeret er dessuten et hjelpeorgan for leger som har risikopersoner til kontroll eller polyposepasienter til behandling.
3.4.3.6 Foreninger og grupper for pasienter med sjeldne sykdommer
Det finnes enkelte pasientforeninger/grupper for pasienter med sjeldne tilstander med kreft som delsymptom. Grupper finnes for ataxia telangiectasia (AT), nevrofibromatose (NF) og tuberøs sklerose (TS). Det finnes planer om å starte en forening for familier med von Hippel-Lindau sykdom. Formålet med slike grupper er generelt å støtte hverandre og å informere familie, omgivelser og medlemmer om forskjellige tiltak som kan bedre dagliglivet. Foreningene er erfaringsmessig av stor betydning for de som er rammet av tilstanden. Dessuten bidrar de til å skaffe informasjon om tilstandene.
I noen av foreningene finnes det medisinske fagråd som kan være behjelpelige med videreformidling til undersøkelse. Det kan variere noe hvor godt fagrådene fungerer; blant annet kan det være vanskelig å oppnå kontakt med leger i fagråd på grunn av stort arbeidspress.
Fotnoter
Tallene som er brukt i punkt 3.1 er i hovedsak hentet fra Kreftregisteret: Kreft i Norge 1996 (Oslo, 1999)
Norsk Bryst Cancer Gruppe: Brystkreft. Diagnostikk og behandling. En veiledning (5. utgave, 1998)
J. T. Wijnen et.al.: Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. New England Journal of Medicine 339:511-518 (1998)
Etter F. C. Fraser: Genetic Counseling. I: American Journal of Human Genetics 26:636-59 (1974)
Norsk Bryst Cancer Gruppe: Brystkreft. Diagnostikk og behandling. En veiledning (5. utgave, 1998)
Pål Møller et.al.: Prospective findings in breast cancer kindreds: annual incidence rates according to age, stage at diagnosis, mean sojourn time, and incidence rates for contralateral cancer. The Breast 7:55-59 (1998)
Pål Møller et.al.: Inherited breast carcinoma. Prospective findings in 1.194 women at risk. Acta Oncologica 35:7-11 (1996)
Norsk Gastrointestinal Cancer Gruppe (NGICG): Kolorektal cancer og analcancer. En veiledning for leger (1999)
H. F. A. Vasen et.al.: A cost-effectiveness analysis of colorectal screening of hereditary nonpolyposis colorectal carcinoma gene carriers. Cancer 82:1632-1637 (1998)
Norsk gynekologisk forening: Veileder i gynekologisk onkologi (1997)
Clinical Genetics Society: The Genetic Testing of Children (1994). Arbeidsgruppa ble ledet av Angus Clarke.
St. prp. nr. 61 (1997-98): Om Nasjonal kreftplan og plan for utstyrsinvesteringer ved norske sykehus.
A. Cull et.al.: The impact of genetic counselling about breast cancer risk on women’s risk perceptions and levels of distress. British Journal of Cancer 79:501-508 (1999)