6 Gentester ved arvelig disposisjon for kreft
I enkelte deler av denne rapporten, og spesielt i de tekniske delene av dette kapitlet ( punkt 6.3 og deler av punkt 6.5), forekommer en del fagterminologi som kan være vanskelig tilgjengelig for personer uten den relevante faglige bakgrunnskunnskapen. Utvalget har funnet dette nødvendig for å sikre at rapporten avspeiler kompliserte biologiske sammenhenger og at fagmiljøene har utbytte av å lese den. Det henvises til annen litteratur for utdypende lesning; her angis kun noen få selekterte referanser. En del faguttrykk står forklart i vedlegg 1. Vi har forsøkt å gjøre hoveddelen av rapporten allment tilgjengelig, slik at gjennomlesing vil gi en generell forståelse.
6.1 Innledning
Med gentest eller genetisk test forstås analyser av DNA, RNA, kromosomer, og/eller visse proteiner og metabolitter, med sikte på å oppdage genfeil som disponerer for arvelig sykdom. Gentester brukes i medisinsk-genetisk sammenheng til å gi en mer presis risikoangivelse og diagnose enn det andre opplysninger (for eksempel slektshistorie) kan gi. Med gentest for arvelig kreft forstås her primært analyser av DNA og RNA.
Lov om medisinsk bruk av bioteknologi skiller mellom ulike typer genetiske undersøkelser etter fødselen: a) diagnostiske undersøkelser, b) presymptomatiske undersøkelser, c) prediktive undersøkelser og d) undersøkelser for å påvise eller utelukke bærertilstand for arvelige sykdommer (jf. vedlegg 2).
De senere år er det gjort store framskritt i oppdagelsen av nye gener som i endret (mutert) tilstand vil medføre sykdom. Oppdagelsene har raskt blitt fulgt opp av gentester som påviser sykdom. I de aller fleste tilfeller har disse testene blitt tatt i bruk uten at det har vært stilt store spørsmål ved bruken av dem. Etter lov om medisinsk bruk av bioteknologi § 6-1 a kalles dette diagnostiske tester. Å stille diagnose er en standard medisinsk prosedyre. Målet er å stille riktig diagnose og å gjøre det tidlig i sykdomsforløpet, for å sette i verk adekvat behandling. Når det gjelder kreftsykdommene, er grunntanken at tidligere diagnose gir større sjanse for at kreftsvulsten ikke har spredd seg, men kan fjernes uten større inngrep. Tilstanden har da større sjanse for helbredelse, noe som anses som et udiskutabelt gode. Dersom man derimot bare flytter diagnosetidspunktet, men ikke bedrer sjansen for overlevelse, kan det diskuteres om tidlig diagnose er nyttig. Det vil i så fall innebære like mange leveår til sammen, men flere i frykt for sykdom.
For genetiske sykdommer der man ikke har kausal behandling (behandling som er rettet mot grunnprosessen i tilstanden), anses diagnostiske tester for å stille tidlig og spesifikk diagnose likevel som et gode. Ved sykdommer som Duchennes muskeldystrofi eller spinal muskelatrofi – som blant annet medfører langsom motorisk utvikling – er de tidlige symptomene ofte vage, og det er vanskelig og til dels plagsomt å få stilt diagnosen. En sikker diagnose vil bety at tiltak kan settes i gang vesentlig tidligere, og at usikkerheten reduseres. Dessuten vil mulighetene for genetisk veiledning bli bedre for foreldre og for andre slektninger av personer med alvorlige sykdommer (for eksempel tanter av barn med Duchennes muskeldystrofi i familien). Dette gjør at diagnostiske tester er akseptert både av leger og pasientorganisasjonene for de forskjellige sykdommene.
Med presymptomatisk test forstås testing for arveanlegg som alltid vil gi sykdom hvis bæreren bare lever lenge nok. Ved tidspunktet for testen er vedkommende uten symptomer eller tegn på sykdommen. Det beste eksempelet er kanskje Huntingtons sykdom. Tilbudet om presymptomatisk test for denne sykdommen har bare vært benyttet av 10-15% av risikopersonene internasjonalt. Dette skyldes blant annet at man ikke kjenner noen behandling som kan hindre sykdommens utvikling. Opplegget omkring testen og oppfølgingen etterpå har vært tidkrevende og omfattende.
Prediktive gentester omfatter tester for arveanlegg med mindre enn 100% penetranse (gjennomslagskraft). Påvisning av arveanlegget for familiær hyperkolesterolemi gir for eksempel en høy, men ikke absolutt, risiko for hjerteinfarkt i yngre alder. Det er påvist svært mange faktorer som predikerer risiko for hjertekarsykdom. Mange av faktorene er arvelige og kan således prinsipielt bli gjenstand for gentesting. Den individuelle risiko er bestemt av den kombinasjon av faktorer som individet er utsatt for; noen faktorer beskytter og andre disponerer.
Skillet mellom presymptomatisk og prediktiv test kan være uklart fordi begrepene avspeiler varierende penetranse.
Arvebærertesting omfatter testing for recessive autosomale og X-bundne arveanlegg, men ikke dominante arveanlegg (slik som presymptomatisk og prediktiv testing). Eksempler er testing av søstre av gutter med Duchennes muskeldystrofi eller Fragilt X-syndrom for å se om det er risiko for at det kan finnes flere tilfeller i familien. Hvis arvebærertesting skal brukes til å slå fast risikoen for at gitte foreldre skal få et barn med en recessiv sykdom, bør begge foreldre teste seg.
Ved presymptomatisk testing, prediktiv testing og arvebærertesting kreves informert samtykke fra den som skal testes. Vedkommende må dessuten være over 16 år, og det skal gis genetisk veiledning før, under og etter prøvetaking. Undersøkelsestyper og -metoder krever godkjenning.
6.2 Kreftrelaterte gener
6.2.1 Generelt
Gener som i endret (mutert) tilstand er vist å bidra til kreftutvikling, kan inndeles i tre grupper: onkogener, tumor-suppressorgener, og reparasjonsgener.
Onkogener er muterte utgaver av proto-onkogener. Proto-onkogener koder for proteiner med normal vekststimulerende evne. I mutert tilstand vil slike gener ofte uttrykke for store mengder protein, noe som gir for mye vekststimulans og derfor kan resultere i eller bidra til ondartet vekst.
Tumor-suppressorgener koder for normale veksthemmende proteiner. Mutasjoner i tumor-suppressorgener kan inaktivere funksjonen og således oppheve den veksthemmende effekten.
Reparasjonsgener koder for proteiner som normalt reparerer ulike skader på arvestoffet. Mutasjoner i slike gener gjør at skader ikke blir reparert og derfor akkumuleres i cellen. Hvis bestemte kreftrelaterte gener skades, og et visst antall slike skader hoper seg opp i cellen, kan dette resultere i eller bidra til ondartet vekst.
Alle tre typer gener kan være mutert i kimbanen, det vil si at den muterte genkopien er arvet fra mor eller far. Individer med slike kimbanemutasjoner er disponert for å få kreft fordi de i utgangspunktet har en mutasjon i alle sine celler. Vi vet imidlertid at det er nødvendig med tilleggsendringer i den enkelte somatiske celle for å initiere og fremme tumorvekst. Anslagsvis er 5-10 somatiske mutasjoner nødvendig for ondartet vekst. Likevel er det sannsynligvis bare visse mutasjoner som har initierende effekt, og analogt er det kun visse endringer som gir en tumor evne til å metastasere (spre seg). Figur 6.1 illustrerer den somatiske mutasjonsteori: utviklingen av en svulst fra en normal celle, via et forstadium (en premalign lesjon) til et ondartet stadium (en malign tumor) og endelig til spredning (en metastase).
Det bemerkes at somatiske mutasjoner (ervervede mutasjoner) kan brukes til tidlig oppdagelse av ny lokal tumor, til identifikasjon av mikrometastaser, til differensialdiagnose, til prognosevurdering og eventuelt til behandlingsvalg.
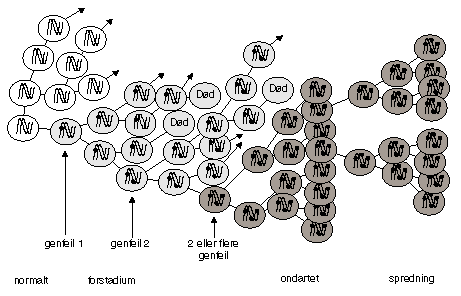
Figur 6.1 Monoklonal utvikling av en svulst
6.2.2 Kreftrelaterte gener spesielt
Oppdagelsen og utviklingen av tester for mutasjoner i bryst- og eggstokkreft-genene, BRCA1 og BRCA2, og genene MSH2 og MLH1 for arvelig non-polypøs tykk- og endetarmkreft (HNPCC), er blitt gjenstand for omfattende debatt og interesse. Særlig gjelder dette BRCA1- og BRCA2-genene. Testing for disse genene er omtalt i punkt 6.4.1. Tabell 6.1 gir en oversikt over andre kreftrelaterte gener som kan være aktuelle for testing, og hvilke det utføres testing for i Norge i dag.
6.3 Metoder som brukes til gentesting i dag
Det er et begrenset antall kreftsykdommer det i dag utføres gentester for, både nasjonalt og internasjonalt. Dette avspeiler først og fremst vår begrensede kunnskap om sykdommene, de disponerende genene og deres funksjonelle aktivitet. Men det sier også noe om begrensningene i teknologien. Dette synliggjøres blant annet i det faktum at diagnostisk gentesting mange steder foregår i nært samarbeid med forskningslaboratorier. Mange av de aktuelle gentester krever spesiell ekspertise for gjennomføring og for tolkning av resultatene. Dessuten har mange av metodene ikke tilstrekkelig prediktiv verdi hver for seg. Derfor brukes ofte et sett av metoder. Både de biologiske og teknologiske begrensningene gjør det derfor særdeles viktig at klinikk, medisinsk-genetisk miljø og det diagnostiske laboratoriet samarbeider, for å oppnå best mulige prosedyrer, vurderinger av resultater og konsekvenser av disse.
6.3.1 Polymerasekjedereaksjon
Generelt er de fleste metoder som brukes ved gentesting, basert på PCR (polymerasekjedereaksjon), publisert som prinsipp av Kjell Kleppe og medarbeidere i 1971 1 og første gang brukt i diagnostisk sammenheng av Saiki og medarbeidere i 1985. 2 Ved bruk av en enzymatisk reaksjon (skjematisk framstilt i figur 6.2), mangfoldiggjøres (amplifiseres) DNA-templat (mors og fars gensett) i så mange kopier at man kan synliggjøre en genfeil ved en mutasjonsanalyse.
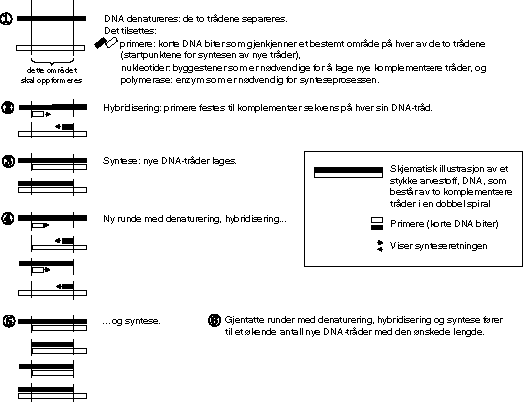
Figur 6.2 Polymerasekjedereaksjon. Prinsipp: et bestemt stykke DNA mangfoldiggjøres
6.3.2 Identifisering av mutasjoner
Valget av egnet teknologi er i første omgang avhengig av om det rekvireres undersøkelse av en bestemt (kjent) mutasjon i et gen eller om man ønsker å finne en ukjent mutasjon, såkalt screening av hele genet.
Laboratorieanalysen starter med at DNA eller RNA fra en blodprøve og/eller en vevsprøve isoleres. DNA er stabilt i hele celler, og en blodprøve kan derfor sendes i posten. En vevsbit bør fryses og lagres ved -20° C før DNA-ekstraksjon. DNA frigjøres etter protein-nedbryting og kan deretter isoleres. DNA er det totale arvestoff, og både kodende og ikke-kodende sekvenser (henholdsvis eksoner og introner) er derfor tilgjengelig for analyser. RNA er ustabilt, og prøvemateriale bør derfor fryses umiddelbart og lagres ved – 80° C eller i flytende nitrogen. Man kan eventuelt dyrke cellene i en korttidskultur og deretter isolere RNA. Det kodende mRNA kan analyseres direkte, men ofte omdannes dette ved hjelp av enzymet revers transkriptase til såkalt komplementært DNA (cDNA) før analyse. Ved dannelsen av mRNA er intronene fjernet, og cDNA utgjør derfor kun eksonene. Fordi hele den kodende informasjonen er samlet i et kortere stykke sekvens i cDNA enn i genomisk DNA, kan mutasjonsanalyser utføres hurtigere på cDNA-materiale. For å finne forandringer i introner som kan endre koden av genet, må man ha genomisk DNA tilgjengelig. Derfor er det ofte nødvendig med tilgang på både DNA og RNA for å identifisere alle mulige mutasjoner.
6.3.2.1 Ukjente mutasjoner
Når man ønsker å finne en ukjent mutasjon i et bestemt gen, kan man i prinsippet sekvensere hele genet. Slike analyser har til nå vært både tid- og kostnadskrevende. Derfor er det utviklet en del screening-metoder som analyserer genet stykkevis og som dermed kan identifisere området hvor mutasjonen ligger. Deretter kan man sekvensere dette området for å påvise den eksakte mutasjon. Slike primære screening-metoder inkluderer denaturerende-gradient-gel-elektroforese (DGGE) og varianter av denne, enkelt-trådig-konformasjons-polymorfisme (SSCP) og varianter av denne, protein-trunkerings-test (PTT), heteroduplexe analyser som inkluderer kjemisk-mismatch-spalting (CMC), endonuklease-mismatch-spalting (EMC) og RNase A spalting, og kløvase-fragment-lengde-polymorfisme (CFLP). 3
Ved søk etter ukjente mutasjoner, vet man verken hvor i genet mutasjonen forekommer eller mutasjonens størrelse. De nevnte metoder er PCR-baserte og identifiserer fortrinnsvis relativt små mutasjoner. Imidlertid er det erfaringsmessig mutasjoner som kun involverer 1-5 baser som forekommer hyppigst. Det vil si at de kvantitativt viktigste årsaker til endring av genfunksjon og utvikling av sykdom, er baseutbytting (missense-mutasjon), baseutbytting som fører til stoppkodon (nonsense-mutasjon) og små delesjoner – tap av baser – eller insersjoner – tillegg av baser – som forandrer genets leseramme (frame-shift-mutasjoner).
Men en del større avvik forekommer og kan forbli uoppdaget hvis man kun bruker PCR-baserte metoder. Derfor benyttes ofte Southern blot-hybridisering for å studere store fragmenter (som oftest i størrelser mellom 400 baser og 40 kilobaser). En slik analyse kan påvise store delesjoner, insersjoner og fragmenter med avvikende størrelse eller intensitet (gendose). Ulike cytogenetiske teknikker vil også påvise store endringer i arvemateriale. Noen av disse vil gi informasjon om hele genomet, mens andre identifiserer utvalgte områder. Slike teknikker brukes relativt lite i forbindelse med testing av arvelig disposisjon for kreft.
6.3.2.2 Kjente mutasjoner
Gentesting kan, som nevnt, enten være å lete etter ukjente mutasjoner ( screene), eller å påvise kjente mutasjoner. Ved påvisning av en kjent mutasjon, må denne tidligere ha vært vist assosiert til den aktuelle sykdom. Den kan ha vært påvist hos andre medlemmer av samme familie, eller den kan forekomme spesielt hyppig i en befolkning. Ofte tilbys mutasjonsanalyser av et visst antall kjente mutasjoner i en befolkning og eventuelt screening av begrensede områder – som man vet er hyppig mutert – i det aktuelle gen.
Kjente mutasjoner blir ofte identifisert ved hjelp av direkte sekvensering av det aktuelle PCR-oppformerte fragmentet. Et avvik på én enkelt base sammenliknet med den normale sekvensen, vil da umiddelbart bli oppdaget. Det er også i bruk flere ulike, enkle teknikker som er tilpasset identifikasjon av én bestemt mutasjon. De inkluderer følgende: allel-spesifikk oligonukleotid-hybridisering (ASO), allel-spesifikk PCR (ASP), oligonukleotid-ligering-amplifikasjon (OLA), restriksjons-enzym (RE) -baserte tester, ligase-kjedereaksjon (LIG), og en nylig beskrevet metode kalt multiplex allel-spesifikk diagnostisk assay
(MASDA). 4
I tillegg til de generelle betraktninger som er beskrevet ovenfor, er kunnskap om de enkelte gener og hyppigheten av bestemte mutasjoner med på å influere valget av analyser. Bestemte mutasjoner kan forekomme spesielt hyppig i enkelte populasjoner. Noen gener muterer fortrinnsvis innenfor bestemte områder av genet, og noen gener viser ofte en bestemt type mutasjon. For eksempel muteres genet for polypose, APC, i mer enn 80% av tilfellene i en såkalt mutasjon-kluster-region i siste ekson. TP53-genet, som er mutert i mer enn 50% av alle solide svulster og i kimbanen hos Li-Fraumeni-pasienter, muteres i mer enn 80% innenfor eksonene 5-8. Flere gener har ofte mutasjoner som fører til endring i størrelsen av proteinet, og PTT kan derfor være et godt valg som første screening-metode.
Diagnostiske prosedyrer inneholder gjerne en prioriteringsrekkefølge av metoder, tilpasset det man vet om den enkelte pasient og vedkommendes familiehistorie og kunnskap om genet eller genene som skal undersøkes.
6.4 Arvelige kreftsykdommer
Mye av forskningen rettet mot arvelig kreft er utført på de sjeldne kreftsykdommene. Denne kunnskapen har så blitt overført til studier av de store kreftformene, som nå er i fokus for den internasjonale forskning. De arvelige kreftsykdommene vi kjenner i dag, utgjør ca. 5% av all bryst- og eggstokkreft og ca. 5% av all tykk- og endetarmkreft. Dette gjør at et relativt stort antall familier er aktuelle for gentesting. Gentesting utføres i dag både for noen sjeldne kreftsyndromer og for noen av de vanlige sykdommene.
I tabell 6.1 er de fleste kreftsykdommer og -syndromer med kjent arvelig disposisjon listet opp. Tabellen anslår også frekvensen av de ulike sykdommene og antallet personer som kan være aktuelle for gentesting. Dessuten er det angitt hvorvidt og i så fall hvor i Norge det gjøres gentester for de enkelte sykdommer.
Punktene 6.4.1-6.4.6 beskriver i mer detalj noen av de kreftsykdommer som er listet i tabellen.
Bruksanvisning for tabell 6.1
Antallet Klinisk syke er basert på antakelser om startalder, dødelighet og sykdomsvarighet. For en del av de sjeldne tilstandene er det oppgitt kjente tall.
Tallet i kolonnen Uaffiserte genbærere er basert på startalder og er ment å gi et inntrykk av størrelsen på det aktuelle kontrollprogrammet.
I kolonnen Aktuelle for gentesting har vi forsøkt å gi en vurdering av de ulike aktuelle gentestenes nytteverdi etter følgende skala:
- +++
gjøres og antas å være nyttige etter vanlige kriterier;
- ++
gjøres og antas å være nyttige, men det er usikkerhet med hensyn til flere faktorer;
- +
gjøres, men det er usikkerhet med hensyn til flere faktorer;
- 0
liten klinisk nytte påvist hittil;
- -
neppe grunn til gentester den i norsk befolkning.
Kriteriene som ligger til grunn for vurderingene, er blant annet diskutert i punktene 4.3 og 7.2.1.1-5. De viktigste er følgende:
Testen har tilstrekkelig klinisk spesifisitet og sensitivitet. En test som påviser mutasjoner hos mer enn halvparten av de som faktisk har feil i genet, men ikke hos de som ikke har slike feil, vil for eksempel tilfredsstille dette kriteriet.
Testresultatet får følger for oppfølging/ behandling (tidlig diagnose vites eller antas å bedre livsutsiktene).
Kostnadene ved testen er akseptable.
Testen er tatt i bruk i land det er naturlig å sammenlikne seg med.
Pasientorganisasjoner og liknende ser et behov for bruk av gentesten.
Tester der alle disse kriteriene er oppfylt, er anført med +++ i tabellen. Bryst-/eggstokkreft, brystkreft, familiær adenomatøs polypose (FAP) og von Hippel-Lindau er eksempler på tilstander der gentesting er tatt i bruk og akseptert fordi testen gir klinisk nyttig informasjon som ikke kan skaffes på annet vis.
++ er angitt for tester hvis kliniske nytte er mindre åpenbar og prognosen til identifiserte genbærere mindre kjent. Slike tester gjøres et begrenset antall steder.
Tallene i parentes angir hvor mange nordmenn det kan være aktuelt å teste for den enkelte tilstanden. En nordmann på 40 år har gjennomsnittlig 2 barn selv og en bror eller søster med 2 barn. Ved 60 års alder har han eller hun i tillegg 4 barnebarn og søskens barn og barnebarn. Foreldre, barn og søsken har a priori 50% risiko, barnebarn/nieser/nevøer har 25% risiko. Basert på Huntingtons sykdomstall med startalder 40 år er det omkring 5 personer det kan være aktuelt å teste i en slik familie. Det kan være aktuelt å teste flere lenger ut i familien på fars eller mors side avhengig av tilstanden og familiens sykdomshistorie.
Kolonnen Gentest i Norge/sted angir hvilke analyser som utføres i Norge, og eventuelt sted: US (Ullevål sykehus), DNR (Det Norske Radiumhospital), HS (Haukeland sykehus) eller RiTø (Regionsykehuset i Tromsø). Koplingsanalyser er ikke regnet som gentest, og en gentest betyr ikke nødvendigvis en analyse av hele genet.
Tabell 6.1 Arvelige dominante kreftsykdommer/syndromer og recessive syndromer med kreft som et framtredende trekk
| Sykdom | Gen | Angitt frekvens | Klinisk syke | Start-alder (snitt) | Uaffiserte genbærere | Aktuelle for gentesting | Vanligste svulstform/lokalisasjon | Gentest i Norge/sted |
| Dominant arvelige kreftsykdommer/-syndromer: | ||||||||
| Bryst-/eggstokkreft | BRCA1 | 1:1.000 | 500 | 50 | 5.000 | +++ (10.000) | Bryst, eggstokk | US, DNR, HS |
| Brystkreft | BRCA2 | 1:1.000 | 500 | 50 | 1.250 | +++ (10.000) | Bryst (også hos menn) | US, DNR |
| Familiært melanom | CDKN2A | 1:1.000-2.000? | 1.000? | 40 | 2.000? | 0 (5.000?) | Malignt melanom, buk spytt-kjertel, dysplastiske nævi | |
| Hereditær ikke-polypøs kolorektal cancer (HNPCC) | MSH2, MLH1 | 1:3.000 | 250-300 | 50 | 800 | ++ (5.000-6.000) | Tykk- og endetarm, magesekk, livmorslimhinne | DNR |
| Familiær adenomatøs polypose (FAP) | APC | 1:5.000-10.000 | 300-500 | 20 | 120-150 | +++ (1.000) | Tykk- og endetarm, magesekk, livmor-slimhinne | US, RiTø |
| Retinoblastom | RB1 | 1:10.000-20.000 | <50 | 1 | - | ++ (<200) | Retinoblastom, osteosarkom | |
| Multippel endokrin neoplasi 2 (MEN2) | RET | 1:30.000 | ~70 | 20-30 | 20-30 | +++ (<100) | Medullært skjoldbrusk kjertelkreft, feokromocytom i binyrer | HS |
| Li-Fraumeni | TP53 | 1:50.000? | <70? | 40? | 40? | ++ (~100) | Sarkom, brystkreft, hjernekreft, leukemi | DNR |
| Multippel endokrin neoplasi (MEN1) | MEN1 | 1:70.000? | ~40? | ~40? | 100? | +++ (300) | Øycelle i bukspytt, biskjoldkjertel, hypofyse Carcinoid | |
| Familiær papillær nyrekreft (HPRC) | MET | 1:100.000? | 10-20 | 40 | ~20 | (<100) | Papillær nyrekreft | |
| Wilms tumor 1 | WT1 | 1:100.000 | <5 | <5 | <5 | ++ (<20) | Nefroblastom (Wilmstumor) | |
| Dominant arvelige syndromer med kreft som del av sykdomsbildet: | ||||||||
| Nevrofibromatose type 1 | NF1 | 1:4.000 | 1.200 | <1 | <1 | - (<10) | Nevrofibromer, nevrosarcomer, gliomer | |
| Tuberøs sklerose 1 | TSC1 | 1:10.000 | 70? | <5 | ? | ++ (200) | Hamartom, angiofi brom, angiomyolipom i nyre | |
| Tuberøs sklerose 2 | TSC2 | 70? | <5 | ? | ++ (200) | |||
| Beckwith-Wiedemanns syndrom (BWS) | 1:15.000 | -100 | <1 | Få | - | Wilms tumor, div. andre svulster | ||
| Von Hippel-Lindau (VHL) | VHL | 1:36.000 | -70 | <15 | <15 | +++ (200) | Nyrekreft (åresvulster i netthinne og sentral nervesystem), feokromocytom | |
| Nevrofibromatose type 2 | NF2 | 1:40.000 | -50 | <20 | <20 | ++ (50) | Vestibulære schwannomer, meningiomer, gliomer | |
| Multiple eksostoser | EXT1, EXT2, EXT3 | 1:50.000 | 50-100 | <10 | ? | - | Eksostoser, sjelden brusksarkom | |
| Gorlins syndrom | PTCH | 1:56.000 | <10 | 20 | ? | - | Hudkreft | |
| Peutz-Jegher syndrom | LKB1 | 1:50.000-100.000 | <50 | <5 | Få | ++ (<10) | Kreft i bryst og skjoldbruskkjertel, hamartomer, magesekk, bryst | |
| Cowden syndrom | PTEN | 1:500.000-1.000.000 | <10 | ? | Få | ++ (?) | Multiple hamartomer (hud, slimhinner, bryst, thyroidea, nyre) | |
| Recessivt arvelige syndromer med kreft som del av sykdomsbildet: | ||||||||
| Fanconis anemi | FANCC, FANCA | 1:100.000? | <10 | <5 | 1:300 | - | Myelogen leukemi, pancytopeni, div. svulster | |
| Ataxia teleangiectasia (AT) | ATM | 1:120.000 | 10 | <5 | 22.000 | - | Lymfom, leukemi, brystkreft hos kvinnelige bærere (recessivt arvelig) | DNR |
| WAGR (Wilmstumor, Aniridi, genitale anomalier, retarda sjon) | PTCH | 1:200.000 | <5 | <1 | Få | - | Wilms tumor (nefroblastom) | |
| Xeroderma pigmentosum | XPB, XPD, XPC | 1:250.000? | <5 | <5 | ? | - | Hudkreft | |
| Bloom syndrom | BLM | 1:1.000.000? | <5 | <5 | ? | - | Leukemi, div. solide svulster |
6.4.1 Brystkreft
Det har lenge vært kjent at bryst- og eggstokkreft i noen tilfeller opptrer i familier og har sitt grunnlag i arv (ca. 5% av alle tilfeller skyldes dominant arv). Nære slektninger av syke har økt risiko for å få sykdommen. Dersom sykdommen har debutert hos en pasient før fylte 40 år, er risikoen for en søster angitt til anslagsvis 3 ganger høyere enn bakgrunnsrisikoen i befolkningen, og den øker til 5 ganger høyere dersom det dreide seg om dobbeltsidig brystkreft.
To gener som gir dominant disposisjon for bryst- og eggstokkreft, er BRCA1 og BRCA2. Mutasjoner i genene gir høy risiko for brystkreft (opp mot 80% livstidsrisiko) og eggstokkreft (40-60% livstidsrisiko) og er til sammen ansvarlig for de fleste av tilfellene av dominant arvelig bryst- og eggstokkreft. Resten antas å skyldes mutasjoner i gener som ennå ikke er identifisert. For mutasjonsbærere av BRCA1/BRCA2 er det sannsynlig at prognosen for brystkreft er relativt god, og at dødeligheten i hovedsak skyldes eggstokkreft.
BRCA1 og BRCA2, lar seg teste for sykdomsdisponerende mutasjoner, men hvert av genene er store, og det er derfor kostbart og tidkrevende å søke gjennom hele genet for å påvise eventuelle mutasjoner. Alternativt kan man teste for kjente mutasjoner (for eksempel alle man har funnet i Norge), men siden mange av mutasjonene bare finnes i enkelte familier, vil ikke alle genfeil påvises ved denne tilnærmingen.
BRCA1 gir også en liten økning av risikoen for prostatakreft og tykk- og endetarmkreft. Bærerfrekvensen (andelen i befolkningen som har mutasjon i genet) for BRCA1 er anslagsvis 1/1.000.
Foundermutasjoner (grunnleggermutasjoner)
I Norge utgjør sannsynligvis noen få (mindre enn 5) forskjellige mutasjoner en stor del (over 50%) av alle BRCA1-mutasjoner. Dette er såkalte foundermutasjoner. De er en følge av den populasjonsgenetiske flaskehals som svartedauden representerte. De sykdomsgener som slapp gjennom svartedauden med genetiske foundere (grunnleggere), har siden ekspandert med en meget sterk befolkningsøkning, fra 150.000 til 10 millioner (inkludert etterkommere av utvandrerne til USA). Muterte BRCA1-gener har vært blant disse. De har i særlig grad nådd en høy frekvens på Sørvestlandet og forklarer en høy frekvens av bryst- og eggstokkreft der.
Dette gjør det mulig å finne en meget stor andel av alle BRCA1-mutasjoner ved å lete etter disse få, spesifikke mutasjonene. For eksempel kan det om ønskelig søkes etter mutasjoner hos alle med brystkreft, eller i hele grupper av kvinner i en bestemt geografisk region, og ikke bare i familier som har høy risiko på bakgrunn av slektstavlen.
6.4.2 Arvelig spesifikk brystkreft og spesifikk eggstokkreft?
Man kjenner ikke til om det finnes ren arvelig brystkreft eller ren arvelig eggstokkreft av noe vesentlig omfang. Det er ikke isolert gener som gir slik kreft. Kreftgener pleier, så vidt man vet, å gi sykdom i flere organer. De gener som til nå er kjent, og som er ansvarlig for hovedtyngden av arvelig bryst- og eggstokkreft ( BRCA1 og BRCA2), gir høy risiko for både bryst- og eggstokkreft. I små familier hender det at den ene eller andre av disse sykdommene ikke forekommer, men det er trolig på grunn av tilfeldigheter.
6.4.3 Tykk- og endetarmkreft
Man antar at de fleste tilfeller av tykk- og endetarmkreft oppstår fra godartede svulster (polyp-per/adenomer). Antakelsen baserer seg på at
forekomsten av polypøst vev i tidlige kreft-stadier er høy;
forekomsten av malignitet (ondartethet) i polyppene øker med størrelse, dysplasi (vekstforstyrrelse) og villøs arkitektur (morfologisk endring i tumor som minner om fingerliknende strukturer);
de geografiske høy- og lavinsidens-områder for polypper og kreft er sammenfallende;
fordelingen i de ulike segmenter av kolon er den samme for polypper og kreft, med samme aldersforandring;
pasienter med familiær adenomatøs polypose (FAP) kan ha hundre til tusentalls av polypper og har nesten alltid kreftutvikling før 50 års alder; og
fjerning av polypper forebygger kreftutvikling.
Generelt øker adenomforekomsten i tarmen med alder. Adenomer forekommer sjelden før 50 års alder. I Norge er prevalensen ca. 25% mellom 50 og 60 år, og rundt 50% i alder over 70 år. Ut fra prevalensen av adenomer og insidensen av kreft, antar man at kun 5-10% av alle adenomer blir til kreft hvis de får stå.
Genetiske faktorer er viktige i utviklingen av tykk- og endetarmkreft. Flere faktorer som gir økt risiko for tykk- og endetarmkreft, er kjent, slik som visse kostbestandeler og betennelsestilstander i tarmen. Det antas at mindre enn 5% av tilfellene av tykk- og endetarmkreft oppstår hos personer med kjent dominant arvelig predisposisjon for sykdommen. De to syndromene som utgjør størst andel av disse, er familiær adenomatøs polypose (FAP) og arvelig non-polypøs tykk- og endetarmkreft (HNPCC). Genetisk testing er mulig for begge disse syndromene, og det er etablert kontrollopplegg for friske med høy risiko for FAP eller HNPCC.
Vi kjenner til et antall arvelige kreftsykdommer med tykk- og endetarmkreft som en del av sykdomsbildet. I tillegg til de kjente arvelig sykdommene, forekommer familiær opphoping av tykk- og endetarmkreft uten at de ansvarlige arveanlegg hittil er identifisert (jf. figur 3.2).
6.4.3.1 HNPCC
HNPCC utgjør omlag 1% av arvelig tykk- og endetarmkreft. HNPCC følger autosomal dominant arvegang, og barn av HNPCC-pasienter har 50% risiko for å arve det muterte genet. HNPCC inndeles i Lynch syndrom 1 og 2. Type 2 er karakterisert ved at det oppstår kreft også i organer utenfor tykk- og endetarmen. Sykdomsdebut er som regel etter fertil alder, og hos unge menn er første sykdomstegn polypp eller kreft i tykk- eller endetarm, mens det hos kvinner ofte er endometriecancer (kreft i livmorens slimhinne). Andre typer ekstra-koloniske svulster (svulster utenfor tarmen) som kan forekomme, er svulster i øvre gastrointestinal traktus, inkludert bukspyttkjertel og galleganger, i eggstokker, i urinveier, i hjernen og i huden.
Den arvelige disposisjonen for HNPCC skyldes mutasjon i ett av flere gener som koder for proteiner i ett av kroppens reparasjonssystemer, den såkalte mismatch-reparasjonen. Defekt i ett av disse genene finnes i en stor andel av HNPCC-familiene. Det er påvist kimbanemutasjoner i fem av disse genene, men to av dem, MSH2 og MLH1, utgjør majoriteten av alle HNPCC tilfeller (anslått til mer enn 80%). Svulstene fra HNPCC pasienter kjennetegnes av såkalt genomisk ustabilitet – små feil mange steder i arvematerialet på grunn av defekt reparasjon. Mønsteret av slike feil kan antyde hvilket gen som bør analyseres og kanskje hvor i genet mutasjonen mest sannsynlig sitter, hvis man skal lete etter en ukjent mutasjon.
I familier hvor mutasjonene er kjente kan man tilby gentesting for risikopersoner før livslang kontroll med koloskopi og ultralydundersøkelser av livmoren igangsettes.
6.4.3.2 FAP
Familiær adenomatøs polypose (FAP) utgjør mindre enn 1% av arvelig tykk- og endetarmkreft og kjennetegnes blant annet ved hundrevis av polypper i tarmen og debut i barnealder. Dersom tykktarmen ikke fjernes profylaktisk, vil det med tida utvikle seg kreft i noen av disse polyppene. Arvegangen for FAP er autosomal dominant, og barn og søsken av syke har nær 50% risiko for kreft. I Norge er det omlag 100 familier med dette arveanlegget. Totalt sett vil omtrent én per 10.000 fødte i Norge utvikle FAP. Bakgrunnsrisikoen i normalbefolkningen for å utvikle polypose er meget lav. Likevel er det slik at gjennomsnittlig ett menneske hvert år vil utvikle polypose uten at sykdommen finnes i familien fra før. I slike tilfeller har en ny kimbanemutasjon oppstått i det disponerende genet, APC.
Polyposen utvikler seg hos de fleste i alderen mellom 10 og 20 år. Hos noen debuterer den noe senere, men sjelden etter 40 års-alderen. Polypose er ikke bare en tarmsykdom; de fleste av pasientene får små fortetninger i knokkelvevet i underkjeven, og noen får også feil ved tannanleggene. Hos over 90% av arveanleggets bærere oppstår det i fosterlivet noen små mørke pigmentflekker på netthinnen. Flekkene er medfødt, helt harmløse og gir ingen symptomer. Når flekkene er tilstede, er dette et prognostisk tegn på at sykdommen vil utvikle seg. Endringene på netthinna er vanskelige å oppdage, men kan påvises av øyelege.
Det finnes etterhvert en del rapporter som tyder på at personer med FAP kan ha en noe økt tendens til utvikling av ondartede svulster i andre organer enn tarm. Blant annet er det rapportert slike svulster i lever, gallegang, binyre og skjoldbruskkjertel. I henhold til dagens viten er risikoen liten; kun få slike tilfeller er rapportert i Norge.
Bærertilstand for FAP kan påvises uten gentest, men skyldes mutasjon i genet APC som normalt koder for proteiner med svulstundertrykkende effekter. I mutert tilstand i kimbanen disponerer genet for multiple polypper i tarm og for kreft i ung alder. Mutasjoner forekommer fortrinnsvis i en bestemt del av genet, og de fleste mutasjoner medfører at det dannes et for kort protein. Sammenhenger mellom bestemte mutasjoner og fenotype er vist.
6.4.4 Retinoblastom
Retinoblastom er en ondartet svulst på netthinnen i øyet som forekommer hos 1 av mellom 10.000 og 20.000, det vil si 3-6 rammede barn per år i Norge. Krefttypen i øyet er av og til medfødt og debuterer oftest før 6 år (retinaceller opphører å dele seg ved denne alder). Det forekommer såvel ensidige som dobbeltsidige svulster. Pasienter med to svulster har tidligere sykdomsstart enn de med én, gjennomsnittlig 12 mot 18 måneder. Genet som kan disponere for sykdommen, RB1, er isolert. Det koder for et protein som normalt har en svulstundertrykkende (tumor-suppressor) funksjon. De dobbeltsidige svulstene skyldes én kimbanemutasjon i RB1, én somatisk mutasjon i RB1 og eventuelt andre somatiske mutasjoner. De ensidige skyldes som oftest to somatiske RB1-mutasjoner (og eventuelt andre) i øyebunnen, men omkring 1/7 av disse har også en kimbanemutasjon.
Pasienter med retinoblastom har også økt risiko for benkreft, spesielt i armer og ben, men også i resten av skjelettet. Denne opptrer ofte i bestrålte felt, men forekommer også uavhengig av strålebehandling. Pasientene har i tillegg økt risiko for ondartede bindevevssvulster, og det kan også sees svulster i bakre del av storehjernen, pinealomer.
Fordi behandlingen er blitt bedre, vil personer med retinoblastom kunne overleve til voksen alder og selv få barn med samme sykdom. Hvert enkelt barn av de som har en RB1-kimbanemutasjon, har en estimert risiko på 45%. Man må derfor ha et langtidsopplegg for oppfølging. Det er ikke identifisert kimbanemutasjoner i RB1 hos norske retinoblastompasienter, og de mutasjoner som er kjent fra litteraturen forekommer på ulike steder i genet. Ulike typer mutasjoner er beskrevet. Dette, og det faktum at genet er relativt stort, utgjør grunnen til at mutasjonsanalyser ikke tilbys i Norge.
6.4.5 Nevrofibromatose type 1
Nevrofibromatose type 1, eller von Recklinghausens sykdom, forekommer med en hyppighet på 1:4.000 eller ca. 1.200 syke i Norge, det vil si en sykdomshyppighet på ca. 15 tilfeller per år. Tilstanden gir manifestasjoner i hud, nervesystem og skjelett, og karakteristisk opptrer en rekke godartede svulster i hud og nervesystem. Disse pasientene har en generell økt risiko (i størrelsesorden 5%) for kreft sammenliknet med normalbefolkningen, og det forekommer særlig i barnealder nevrofibrosarkomer, Wilms tumor (nefroblastom) og astrocytomer (svulst i nervesystemet). Halvparten av tilfellene forekommer som nye kimbanemutasjoner i NF1-genet. Dette genet koder for et protein som normalt har en svulstundertrykkende funksjon. Det foreligger ingen informasjon som gir sikker sammenheng mellom visse mutasjoner og kreftrisiko. Selv innenfor en familie kan samme mutasjon gi forskjellig fenotype. NF1-genet er svært stort og testes kun i forskningssammenheng. Ingen spesiell oppfølging av pasientene med tanke på kreft er satt i verk, diagnosen stilles ofte sent, og når svulster forekommer, er det ofte umulig å behandle dem effektivt.
6.5 Framtidig gentesting
6.5.1 Videreutvikling av etablerte tester
Testing for tilstedeværelse eller fravær av en bestemt kjent mutasjon, kan gjøres tilfredsstillende med dagens metoder. Det blir i dette henseendet mer et spørsmål om ressurser og kostnader for å dekke det behov som finnes. Imidlertid kan man tenke seg å tilrettelegge ulike effektiviseringstiltak for de enkelte analyser. Testene har verken falske positive eller falske negative resultater. Hovedproblemet er biologisk: man kjenner ikke sammenhengen mellom genotype og fenotype for hver mutasjon. For foundermutasjonene er en i ferd med å skaffe seg god informasjon om dette, siden disse finnes hos mange pasienter. For de private mutasjonene blir det knapt mulig å få slik informasjon. Dette begrenser utsagnskraften av en test betydelig.
Screening etter ukjente mutasjoner er en stor utfordring, blant annet fordi metodene ikke er optimale. En optimal metode for mutasjons- screening bør være 100% effektiv (ingen falske negative eller positive); kilobaselengder av DNA må kunne analyseres allerhelst i ett trinn eller eventuelt i få trinn; man må ikke trenge komplisert utstyr og man må ikke være nødt til å bruke toksiske reagenser; og endelig bør tida operatøren bruker på testen være begrenset og kostnadene lave. Selv om ingen av metodene som brukes i dag er 100% effektive, har de enkelte metoder blitt utviklet og forbedret mye siden de først ble introdusert. Videre optimalisering av de etablerte metodene (jf. punkt 6.3) pågår kontinuerlig i mange laboratorier over hele verden, og disse vil i den nære framtid være de redskapene som anvendes for diagnostisk gentesting.
6.5.2 Det humane genom-prosjekt
Det humane genom-prosjekt (HGP) avsluttet i 1998 en 5 års plan, og lå da foran målsettingene fra 1993. Til nå er 6% av det humane genom sekvensert. I tillegg finnes det 18 eksempler på fullstendig genomsekvens av enklere organismer, inkludert bakterien E. coli og gjær. Det integrerte genetiske og fysiske kart av det humane genom inkluderer data fra mer enn 41.000 cDNA baserte sequence tagged sites (STS), som representerer over 30.000 humane gener. 5 De kromosomale lokalisasjoner av disse genene viser at gentettheten er større på enkelte kromosomer enn på andre. Det er antatt at det humane genom består av i størrelsesorden 60.000-100.000 gener. I så fall vil de 30.000 genmarkørene representere nær 50% av alle humane gener. Imidlertid vil den framtidige totale humane sekvens gi innsikt i om dette anslåtte antall er for lavt; for eksempel hvor utbredt nested genes (gener inni gener) er. Dette er et fenomen som allerede er kjent for noen humane gener, eksempelvis NF1-genet. I de 30.000 genmarkørene er ca. 4.500 av 6.000 gener med kjent funksjon inkludert.
En av hovedverdiene med et tett og integrert genetisk-fysisk kart over de humane gener, er å akselerere identifikasjonen av humane sykdomsgener, deriblant kreftgener.
Den nye 5 års-planen for HGP, 1998-2003, har åtte målsettinger 6 som også inkluderer den nære og fjernere framtidige storskala gentesting og dens konsekvenser:
Oppnå en høykvalitetssekvens av hele det humane genom i år 2003 (for øvrig 50 års jubileet for oppdagelsen av den doble DNA helixen 7 ).
Forbedre og redusere kostnadene av nåværende sekvensteknologi, og utvikle ny teknologi.
Starte identifikasjon av den naturlige sekvensvariasjon som finnes i genomet.
Forbedre og utvikle ny teknologi for funksjonelle studier.
Videreføre komparative genomstudier. Sammenlikning av genomene til ulike arter, gir innsikt i universale mekanismer og identifiserer eksperimentelle modeller for å studere komplekse prosesser.
Klare målsettinger for programmet ELSI ( ethical, legal, and social implications) som er integrert i HGP.
Videreutvikle bioinformatikk – oppbygging og bruk av ulike databaser og analytiske verktøy.
Foreslå og tilrettelegge tiltak for vitenskapelig personale i kontaktflaten mellom biologi og en rekke andre disipliner (data, ingeniørfag, matematikk, fysikk, kjemi, etc.)
6.5.3 Mikrobrikketeknologi (microarray, chips-technology)
Mikrobrikketeknologien synes å være på full fart inn i molekylærgenetisk forskning og klinisk diagnostikk. Teknologien åpner for muligheten til systematisk å studere DNA- og RNA-variasjon, ved å vurdere multiple komponenter som virker samtidig i de individuelle biologiske prosesser.
I et 60 siders spesialsupplement til januarnummeret av Nature Genetics 1999, omtales mikroarrays og anvendelse av dem i en samling oversiktsartikler. 8 Her finnes informasjon om teknologien for produksjon av mikroarrays og om bruk i forskjellige biologiske problemstillinger.
I 1975 viste Ed Southern at DNA kunne bindes til et fast underlag, og Southern blot kan derfor sies å være det første array. 9 Dette ble videreutviklet til filterbasert screening av nukleinsyrekloner. Pat Brown og kolleger overførte dette prinsippet til et mikrosystem ved å lage mikroarrays på et ikke-porøst fast underlag av glass. De plasserte 10.000 cDNA-kloner med robotisert hjelp på et 2x3 cm glass, og hybridiserte med fluorescens merkede prober. Fargevariasjon kunne så computeranalyseres.
De mest omtalte arrays er oligonukleotid arrays som er designet for å evaluere spesifikke sekvenser, slik som BRCA1- og TP53-genene. Fodor og kolleger ved Affymetrix bruker fotolitografi-basert metodikk for å lage DNA chips med tusenvis av korte kjente oligosekvenser på en silikonoverflate. Dette firmaet har i dag utviklet 20 ulike gen-chips.
Det er nylig også lansert oligoarrays som ikke avleses som et flourescerende mønster, men som elektriske signaler. Når DNA hybridiserer til sin komplementære oligo på arrayet, bindes også et jernholdig molekyl til komplekset som detekteres av elektroder.
Arvelig bryst- og eggstokkreft er en høypenetrant sykdom, og populasjonsbasert mutasjons-screening kan være aktuelt. Genet BRCA1, som disponerer for sykdommen, viser et komplekst mutasjonsspektrum; mer enn 500 forskjellige mutasjoner er beskrevet internasjonalt. Derfor vil screening etter nye mutasjoner innebære analyse av alle mulige forandringer i heterozygot tilstand. Hybridiseringsbasert sekvensanalyse på mikroarrays åpner for slike muligheter. Med bruk av 96.000 oligonukleotider i arrayhybridisering (kombinasjon av gevinst og tap av flourescensintensitet) skulle deteksjon av alle mulige enkeltbasesubstitusjoner, alle enkelt-basepar-insersjoner og alle 1-5 basepar-delesjoner i 3,43 kb av ekson 11 i BRCA1 teoretisk oppnås. 14 av 15 heterozygote mutasjoner ble identifisert. Nylig er det brukt tilsvarende Gene Chips fra Affymetrix for å screene hele kodende sekvenser av BRCA1 og ATM. Imidlertid opererer man med en mutasjons-deteksjons-sensitivitet i størrelsesorden 90%, det vil si at andelen falske negative svar kan være så høy som 10%. Derfor er hybridiseringsbasert mutasjonsanalyse på arrays foreløpig ikke egnet, hvis behovet er å detektere alle ukjente heterozygote mutasjoner med tilnærmet 100% sensitivitet. Men når spesifikke hybridiseringsmønstre blir kjent for et stort antall mutasjoner, så kan tilstedeværelse av disse undersøkes for en mengde prøver samtidig. Det vil si at man tester for en rekke kjente mutasjoner på en gang, men et negativt svar utelukker ikke at det finnes en mutasjon det ikke er testet for.
Det pågår stridigheter og rettssaker vedrørende patentrettigheter både på hardware-chip-teknologien i seg selv og på genene som settes på arrayet. Mye tyder på at dette vil bli langvarige prosesser. Parallelt arbeider offentlig finansiert HGP med ELSI-programmet.
6.5.4 Mikrolaboratorier
Etter at laboratoriet har mottatt en blod- eller vevsprøve må DNA/RNA ekstraheres, og den delen av DNA/RNA som skal analyseres, må velges, oppformeres med PCR og eventuelt sekvenseres. Dette utføres i dag i trinnvise analyser med bruk av flere maskiner på laboratoriebenken. Framtida kan komme til å bringe mikrolaboratorier hvor trinnvise analyser, som i det nevnte eksempelet, utføres i separate rom i små brikker på størrelse med et bankkort. Mulighetene for utvikling av miniatyr-utgaver av kjemisk utstyr kan sammenliknes med den utvikling vi har sett når det gjelder elektroniske maskiner. Denne framtidas type mikrobrikker vil dramatisk redusere lønns- og reagenskostnader, og forbruket av materiale vil bli svært lite. Noen universitetsmiljøer og firmaer arbeider i dag med denne type teknologi. Det er allerede vist at slike mikrolaboratorier fungerer, og framtidas aplikasjonsmuligheter er svært mange. Den prosesseringen av en blodprøve som ble nevnt ovenfor, er nylig utført i en mikrobrikke med en hundrefolds reduksjon i forbruk av DNA sammenliknet med konvensjonell metodikk. 10
Fotnoter
K. Kleppe et.al.: Studies on polynucleotides XCVI: repair replications of short syntetic DNAs as catalysed by DNA polymerases. Journal of Molecular Biology 56:341-361 (1971)
R. K. Saiki et.al.: Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 239:487-491 (1985)
S. E. McKenzie et.al.: Parallel molecular genetic analysis. European Journal of Human Genetics 6:417-429 (1998)
S. E. McKenzie et.al.: Parallel molecular genetic analysis. European Journal of Human Genetics 6:417-429 (1998)
P. Deloukas et. al.: A physical map of 30,000 human genes. Science 282: 744-746 (1998)
F. S. Collins et. al.: New goals for the U.S. Human Genome Project: 1998-2003. Science 282:682-689 (1998)
G. D. Watson & F. H. C. Crick: Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature 171:737-(1953)
The chipping forecast. Nature Genetics 21:1-60 (1999)
E. M. Southern: Detection of specific sequences among DNA fragments separated by gel electrophoresis. Journal of Molecular Biology 98:503-517 (1975)
R. F. Service: Coming soon: the pocket DNA sequencer. Science 282: 399-401 (1998)