Del 3
Gjeldende reguleringer
8 Reguleringer
8.1 Begrepsbruk
8.1.1 Etiske normer, rettsnormer og profesjonsnormer
Utvalget er i mandatet bedt om å ”gi en oversiktlig framstilling av regler og retningslinjer som i dag angår medisinsk forskning som involverer mennesker og humant biologisk materiale, både i Norge og internasjonalt, herunder Norges deltakelse i internasjonale forskningsprogrammer.” Fremstillingen av gjeldende reguleringer har naturligvis stor betydning som grunnlag for utvalgets vurdering av reguleringsbehovet.
Med regulering i denne sammenheng menes alle normer som påvirker og styrer medisinsk og helsefaglig forskning. Det kan i denne forbindelse skilles mellom tre ulike typer normsett:
Etiske normer
Rettsnormer
Profesjonsnormer
Etiske normer refererer til de oppfatninger om rett og galt og godt og ondt som finnes i et samfunn eller en gruppe. Vi kan skille mellom deskriptiv og normativ etikk. Deskriptiv etikk er beskrivelser av hvilke oppfatninger om rett og galt som finnes, mens normativ etikk er teorier om hvordan man skal kunne skille mellom rett og galt og godt og ondt. Etiske oppfatninger og vurderinger kan befestes i mer eller mindre presise atferdsnormer som uttrykker hva som i alminnelighet eller innen visse grupper eller profesjoner, oppfattes som god og forsvarlig atferd på ulike livsområder. Dette kan kalles etiske normer.
Angivelse av idealer og etiske normer kan være nok for å sikre ønsket atferd, men er ofte ikke et tilstrekkelig grunnlag for å sikre en klar og utfyllende regulering av komplekse forhold. I våre dager er det vanlig at samfunnet gjennom Stortinget vedtar lover for å sikre en hensiktsmessig regulering i overensstemmelse med samfunnets verdigrunnlag. Lover håndheves av organer som samfunnet har utpekt for dette formål, i første rekke av domstolene, men også av forvaltningsorganer. Statens helsetilsyn er for eksempel autorisert til å fastsette visse reaksjoner ved overtredelse av helselovgivningen. De sanksjonsmuligheter som følger av lovbestemte forpliktelser og rettigheter, er den viktigste forskjellen mellom rettsnormer og etiske normer. Man kan dermed si at forholdet mellom etikk og jus er at rettsregler opphøyer (”rettsliggjør”) visse normative oppfatninger som samfunnets offisielle og autoritative syn på hva som er akseptabel og ikke-akseptabel handlemåte, i betydningen lovlig og ulovlig.
I tillegg til skrevne lover finnes ulovfestede rettsregler (sedvanerett) som har samme rang som lovregler, men som blir til gjennom en langvarig prosess der sosiale eller profesjonelle normer og oppfatninger på bestemte retts- eller livsområder, eller i visse bransjer eller profesjoner, festner seg og oppfattes som bindende rettsnormer. I vår tid med en aktiv lovgiver er det nokså uvanlig at ulovfestede rettsregler påberopes og anvendes som ledd i rettslig argumentasjon. Men på rettstomme rom, som for eksempel områder innen medisinsk og helsefaglig forskning, kan det være aktuelt å anvende alminnelige ulovfestede prinsipper slik disse har kommet til uttrykk for eksempel i Helsinki-deklarasjonen, som er en viktig profesjonsnorm skrevet av og for leger.
Rettsreglene kan være både nasjonale og internasjonale. Norge deltar i økende grad i internasjonalt samarbeid og har signert og ratifisert en rekke internasjonale konvensjoner som er folkerettslig bindende og dermed av rettslig betydning ved fastlegging av nasjonal rett.
Profesjonsnormer vedtas og håndheves av profesjonene selv, og de gjelder for medlemmene av profesjonen. Normene kjennetegnes ofte ved at de er utformet av en bestemt gruppe med felles interesser, i motsetning til lover som vedtas av en bredt representert nasjonalforsamling. Profesjonsnormer blir enten formulert i egne regelverk eller de utvikles gradvis gjennom praksis som får tilslutning innen fagmiljøet. Profesjonsnormer kan også vedtas av internasjonale faglige organisasjoner hvis retningslinjer aksepteres av nasjonale faglige sammenslutninger. Også profesjonsnormer uttrykker oppfatninger om hva som er rett og galt, eller etisk versus uetisk. Men i motsetning til rettsnormer som er utformet og håndheves av samfunnets autoriserte organer, angir profesjonsnormene profesjonenes eget syn på hva som er rett og galt. Profesjonen kan ha knyttet sanksjoner til overtredelse, men har ikke samme mulighet til å stille makt og tvang bak disse om samfunnet har. Dette betyr likevel ikke at de nødvendigvis er mindre effektive enn samfunnets rettsnormer. Selv om profesjonsnormene ikke er rettsregler som kan kreves håndhevet for domstolene og ikke har den samme rettslige status som lovgivning og internasjonale konvensjoner, kan de likevel ha rettslig betydning som momenter som bør vektlegges ved bestemmelsen av gjeldende rett på et bestemt område. Siden profesjonsnormer kan inngå i bestemmelsen av gjeldende rett, kan det også være et noe uskarpt skille mellom slike normer på den ene siden og nasjonale eller internasjonale rettsregler på den andre.
8.1.2 Forholdet mellom atferdsregler, organisa- toriske regler og saksbehandlingsregler
Det kan skilles mellom atferdsregler, organisatoriske regler og saksbehandlingsregler. At forskningen skal baseres på forsøkspersoners frivillige og informerte samtykke, samt ulike forbud og påbud knyttet til forskning, er eksempler på atferdsregler for forskere. Opprettelse av kontroll- og tilsynsorganer er eksempler på organisatoriske regler. Regler som fastlegger normer for behandling av søknader fra forskere til de regionale forskningsetiske komiteer er eksempler på saksbehandlingsregler.
Det er ingen eksakt grense mellom disse ulike regeltypene, og det er viktig å se sammenhengen mellom dem. Dersom atferdsreglene er kompliserte, uoversiktlige, ikke harmonisert og/eller uklare, stiller det store krav til saksbehandlingen. Et enhetlig og klart regelverk er ofte en forutsetning for etterlevelse og effektiv kontroll, ved at det motvirker vilkårlighet og sikrer forutsigbarhet. Dette bidrar også til at tolkningen ikke blir for skjønnsmessig, noe som forenkler kontrollorganenes arbeid og reduserer deres ansvar. På den annen side er det ofte umulig å detaljregulere ethvert spørsmål på kompliserte områder. Det må som regel utøves en del skjønn. Detaljregulering er heller ikke ønskelig, fordi regelverket bør være fleksibelt på et område der utviklingen går raskt og virksomheten er uensartet, slik som tilfellet er for medisinsk og helsefaglig forskning.
8.1.3 Nasjonale, internasjonale og andre lands reguleringer
8.1.3.1 Nasjonale reguleringer
Norske reguleringer er i vår sammenheng i første rekke lover og profesjonsnormer utarbeidet av norske myndigheter og organisasjoner. I tillegg omfattes internasjonale reguleringer som gjelder som norsk rett, slik som de menneskerettighetskonvensjoner som ved menneskerettsloven er inkorporert i norsk rett.
8.1.3.2 Internasjonale reguleringer
Internasjonale rettsregler kan være konvensjoner som er folkerettslig bindende for stater, og som den enkelte stat forplikter seg til å gjennomføre i nasjonal rett. Internasjonale konvensjoner om menneskerettigheter er eksempler på slike. Internasjonal rett kan også ha utspring i internasjonal sedvanerett eller alminnelige internasjonale rettsgrunnsetninger. 1 I tillegg er det vanlig å ta med resolusjoner og deklarasjoner som ikke er rettslig bindende i seg selv, men som kan være normgivende, avhengig av utbredelse og aksept.
Norsk lovgivning som regulerer medisinsk og helsefaglig forskning, må være i overensstemmelse med de internasjonale forpliktelser Norge har påtatt seg. Konkret betyr det at norske lover på dette området må være i overensstemmelse med Europarådets konvensjon om menneskerettigheter og biomedisin med tilleggsprotokoller (Oviedo-konvensjonen), 2 som Norge er i ferd med å ratifisere, og sannsynligvis vil ha ratifisert innen 2005, samt legemiddeldirektivet og personverndirektivet. Ved utarbeidelsen av nasjonale reguleringer er det derfor viktig å klargjøre hvilke internasjonale rettsregler Norge er eller vil bli forpliktet av i nær fremtid.
Internasjonale konvensjoner gir vanligvis nasjonale myndigheter et visst spillerom ved utarbeidelsen av nasjonale reguleringer, og angir ofte minimumskrav som statene må oppfylle.
Medisinsk forskning på det internasjonale plan har tradisjonelt vært regulert av profesjonene selv, f.eks. Helsinki-deklarasjonen vedtatt av Verdens legeforening, som er en sammenslutning av nasjonale legeforeninger. Dette er internasjonale profesjonsnormer, til forskjell fra internasjonale rettsnormer (folkerett). Det er opp til de nasjonale profesjonsorganer å påse etterlevelse av internasjonale profesjonsnormer.
Utviklingen av internasjonal rett har i de senere år vært påtakelig på området for medisinsk og helsefaglig forskning. Det gjelder både på det globale plan gjennom FN og på det regionale plan gjennom EU og Europarådet. Dette har bakgrunn særlig i to forhold;
økende globalisering og større behov for harmonisering av nasjonale reguleringer, og
økende gjennomslag for og vektlegging av internasjonale menneskerettigheter.
8.1.3.3 Andre lands reguleringer
Andre lands reguleringer er reguleringer i andre land, og har liten eller ingen direkte betydning for reguleringene i Norge. De kan imidlertid være av stor betydning som sammenligningsgrunnlag, spesielt gjelder dette land som det er naturlig å sammenligne oss med, for eksempel Sverige og Danmark. Ofte vil det være naturlig å tilstrebe en viss harmonisering av regelverket av hensyn til blant annet internasjonalt samarbeid.
9 Utviklingstrekk
9.1 Innledning
Mens resultatene av medisinsk forskning kanskje kan kalles ‘the greatest benefit to mankind’, har opparbeidelsen av slik kunnskap ikke alltid skjedd i tråd med samfunnets idealer og normer. 3 Parallelt med den optimistiske fremtidstro forskningen i sin tid skapte, oppsto det derfor også en mistro til selve forskningen.
Systematiske forsøk på mennesker for å øke medisinsk viten var i utgangspunktet et europeisk fenomen som startet i renessansen. 4 På denne tiden var legenes selvpålagte reguleringer allerede godt utviklet og innarbeidet i pakt med det hippokratiske prinsipp fra 430 f.Kr., som gikk ut på å søke de sykes vel og unngå alt som kan skade pasienten. Profesjonsnormene var ikke utformet med tanke på forskning, men det er lite trolig at leger anså seg ubundet av disse i den forstand at det var fritt frem å gjøre hva man ville med mennesker i forskningens navn. Grensene for det akseptable har imidlertid variert både individuelt hos den enkelte forsker og i samfunnet for øvrig. 56
9.2 Nasjonal utvikling
Gerhard Henrik Armauer Hansen (1841-1912) identifiserte i 1873 leprabasillen (Mycobacterium leprae), og var den første som påviste at en bestemt mikroorganisme forårsaker en kronisk infeksjon. I 1879 podet han materiale fra en spedalsk knute inn i øyet på en kvinnelig pasient som led av den glatte form for spedalskhet. Formålet var å undersøke smitteforholdene ved lepra. 7 Armauer Hansen ble tiltalt for dette, og i straffesaken mot ham uttalte retten:
Tiltalte, hvor meget der end efter det Anførte kan tale til Undskyldning for hans Handlemaade fra et lægevidenskabeligt Synspunct ved den af ham brugte Fremgangsmaade saaledes som den af ham med fuldkommen Aabenhed er erkjendt, med Overlæg har benyttet sin Stilling ligeoverfor 1ste Vidne til at påføre hende en Legemslidelse, som han selv erkjender at han ikke kunne gaa ut fra, at hun vilde have samtykket i at underkaste sig, hvis han på Forhaand havde gjorte henne bekjendt dermed. 8
Armauer Hansen ble fradømt stillingen som lege ved pleiestiftelsen i Bergen, men beholdt stillingen som overlege for de spedalske. Dommen vakte stor oppmerksomhet i vitenskapelige kretser og ble til dels sterkt kritisert av forskere, 9 men gikk relativt upåaktet hen i den alminnelige samfunnsdebatt. 10
I et rettshistorisk perspektiv er saken oppsiktsvekkende både nasjonalt og internasjonalt. Domstolen utledet et krav om informert samtykke ved forskning på mennesker ut fra alminnelige strafferettslige regler om legemskrenkelser. Høyesterettsdommer Knut Blom konkluderte i 1973 med at saken mot Armauer Hansen neppe kan ha vært særlig tvilsom hva skyldspørsmålet angår idet ”handlingen innebar en klar legemskrenkelse av pasienten”. 11 Legeprofesjonens støtte til Armauer Hansen og lignende forsøk tyder imidlertid på at et forskningsetisk prinsipp om informert samtykke ikke stod sterkt på slutten av 1800-tallet. 12 Det skulle gå flere tiår før samtykkeregelen var fullt ut akseptert i vitenskapelige kretser både i Norge og i utlandet. 13
Norske domstoler har forøvrig ikke spilt noen nevneverdig rolle i å utvikle reguleringer av medisinsk forskning, slik de for eksempel har gjort det ved utviklingen av den alminnelige (domstolskapte) erstatningsretten. Dette har sammenheng med at svært få saker om forskningsvirksomhet er brakt inn for domstolene. I det store og hele finnes det i Norge få eksempler på at medisinsk forskning er blitt stemplet som uforsvarlig. En regjeringsoppnevnt granskningskommisjon fant ingen holdepunkter for enkelte spesifikt fremsatte påstander om uetisk medisinsk forskning på mennesker i perioden 1945-1975. 14 Den første forskrift om kliniske utprøvninger på legemiddelområdet kom i 1981. Forskriftens § 10 henviser til Helsinki-deklarasjonen. Den første lov om personregistre, der Datatilsynet ble etablert, kom i 1978. Norge etablerte forskningsetiske komiteer i 1985, noe som var forholdsvis sent sammenlignet med andre land. Før den tid hadde lokale komiteer ved enkelte sykehus anvendt Helsinki-deklarasjonens regler på ad hoc basis.
9.3 Internasjonal utvikling
Oppgjøret etter den andre verdenskrig regnes som et tidsskille for reguleringen av medisinsk forskning. 15 I Nürnberg-prosessen dømte Militær Domstol I i saken USA v. Karl Brandt et al. 23 tyske leger for krigsforbrytelser og forbrytelser mot menneskeheten den 19. august 1947. 16 Legene ble funnet skyldige i hensynsløse eksperimenter på fanger, funksjonshemmede og andre, for å teste tålegrenser for nedkjøling, trykk og lignende.
Med utgangspunkt i internasjonale strafferettslige prinsipper utledet domstolen ti prinsipper for medisinsk forskning, den såkalte Nürnberg-kodeksen. 17 Kodeksen kan sies å fusjonere den hippokratiske legeed med alminnelige menneskerettigheter. 18
I 1945 ble De forente nasjoner (FN) etablert, og i 1948 vedtok FNs generalforsamling Verdenserklæringen om menneskerettigheter. Verdenserklæringen var startskuddet for en utvikling av menneskerettigheter som kom til å få stor betydning, også for reguleringen av medisinsk forskning.
Menneskerettighetene bygger på en naturrettslig oppfatning om at alle mennesker er født frie med en iboende verdighet og med de samme grunnleggende rettigheter. Retten til liv, frihet, selvbestemmelse, rettferdig rettergang, privatliv, tanke- og ytringsfrihet, utdanning og helse er sentrale menneskerettigheter. De fremstår som beskyttelse mot makt- og myndighetsmisbruk, og er dermed viktigst som et vern for de svakeste mot de sterkeste. 19
Verdenserklæringen ledet til vedtakelse av flere internasjonale konvensjoner. Konvensjonene viderefører de etiske normene og idealene som rettslige normer, som de ratifiserende stater har forpliktet seg til å etterleve. De fleste stater har nå sluttet seg til FNs konvensjoner om henholdsvis sivile og politiske rettigheter og økonomiske, sosiale og kulturelle rettigheter, begge fra 1966. Ved vedtakelsen av menneskerettsloven av 1999 ble disse, i tillegg til Den europeiske menneskerettighetskonvensjon og Barnekonvensjonen, gitt status som norsk lov, med forrang foran bestemmelser i andre lover ved motstrid.
I 1964 vedtok Verdens legeforening (en sammenslutning av de nasjonale legeforeninger) Helsinki-deklarasjonen som med utgangspunkt i Nürnbergkodeksen fastsetter en rekke prinsipper for god forskning. Foranledningen var enkelttilfeller av misbruk av forskningsdeltakere. 20 Et eksempel er Tuskege-eksperimentet hvor man studerte utviklingen av syfilis hos fargede menn i perioden 1932-1972 uten at deltakerne verken samtykket, fikk informasjon eller tilgang til effektiv behandling. 21 Eksempler på uetisk forskning inkluderte også risikable eksperimenter på utsatte grupper som foreldreløse barn, pasienter, fanger, soldater, psykisk utviklingshemmede og minoritetsgrupper. Disse var lett tilgjengelige, lette å kontrollere og ble ansett for å stå i en slags ”gjeld” til samfunnet. 22
Heller ikke Helsinki-deklarasjonen forhindret uetisk forskning. I 1966 dokumenterte Henry Beecher 22 uetiske forskningsprosjekter der deltakere ikke ble informert om at de ble inkludert i risikable forsøk. 23 Som en reaksjon på flere slike forhold, vedtok Verdens legeforening i 1975 en tilføyelse til Helsinki-deklarasjonen hvor etikkomiteer ble anbefalt opprettet. Ved å kreve forhåndsgodkjenning av forskningsprosjekter skulle overgrep forhindres og god forskning fremmes.
9.4 Økende rettslig regulering
I Norge er det de siste årene vedtatt en rekke lover av betydning for medisinsk forskning. Den omfattende reguleringen har sammenheng både med den generelle samfunnsutvikling her i landet og med økt internasjonalt samarbeid. Europarådets Oviedo-konvensjon av 1997 med tilleggsprotokoller og EUs legemiddeldirektiv av 2001, som begge bygger på Helsinki-deklarasjonen, stiller relativt omfattende og eksplisitte minstekrav til nasjonal regulering av medisinsk forskning.
Formaliseringen av pasienters og forskningsdeltakeres rettigheter ved kliniske og invasive forsøk må sees i sammenheng med utviklingen av personvernet. Personvernet har sitt utgangspunkt i å ville beskytte privatlivets fred og hindre krenkelser av den personlige integritet. 24 I følge personverntankegangen, som har røtter i amerikansk rett, bør individet ha rett til å bestemme hvilke opplysninger om seg selv som det skal gi fra seg, og hvordan disse opplysningene skal brukes. I Norge og Europa forøvrig har man tradisjonelt lagt et noe mindre individualistisk syn til grunn enn i USA. 25 Hos oss er personvernet rettet mot å beskytte den enkelte mot bruk av personopplysninger som kan utsette individet for nærmere bestemte farer, ulemper eller ubehag. 26 Også på dette område er nasjonal lovgivning et utslag av internasjonale reguleringer, særlig EUs personverndirektiv.
En relativt ny form for særregulering gjelder innsamling, lagring og bruk av humant biologisk materiale. I alle de nordiske landene finnes det nå rettslig regulering av såkalte biobanker. Internasjonalt arbeides det også med reguleringer på dette området, senest ved EU- direktivet om behandling av humane vev og celler av 2004, og Europarådets forslag til biobankkonvensjon. På bioteknologiområdet er FN-organet UNESCOs erklæring om det humane genom og menneskerettigheter av 1997, som Norge har sluttet seg til, av stor betydning.
10 Oversikt over gjeldende reguleringer
10.1 Et fragmentert regelverk
Det finnes i dag ingen egen norsk lov som regulerer medisinsk og helsefaglig forskning i Norge. Reguleringen av denne forskningen består av ulike ”biter” som er spredt i mange ulike regelsett, både nasjonale og internasjonale, som direkte eller indirekte berører ulike sider av medisinsk og helsefaglig forskning.
I det følgende vil enkelte lover, profesjonsnormer og annet materiale av særlig interesse kort introduseres, jf. også tabell 10.1. I kapitlene 11-18 gis det deretter en redegjørelse for gjeldende rett på utvalgte rettsområder.
Tabell 10.1 Oversikt over relevante reguleringer for medisinsk og helsefaglig forskning.
| Regulering | Formål | Virkeområde | Hvordan berøres forskningen? | Bemerkninger |
|---|---|---|---|---|
| Alminnelig forvaltningsrett; forvaltnings-loven, offentlighetsloven | Sikre forsvarlig forvaltning | Offentlige organer og deres ansatte. Gir private enkelte rettigheter | Setter opp saksbehandlingsregler, som vil gjelde for offentlig ansatte forskere samt offentlige organer som fører kontroll og tilsyn med forskning | Regler om habilitet, taushetsplikt, veiledningsplikt, klage, osv. Reglene er generelle og ofte finnes det spesial lover som inneholder mer spesifikke regler på området |
| Menneskerettighetsloven med vedlegg | Sikre respekt og beskyttelse for borgernes grunn-leggende rettigheter og friheter | Offentlige organer og deres ansatte. Indivi-dene har rettigheter og staten har plikter. Staten er folkerettslig forpliktet | Sikrer den enkeltes selv-bestemmelsesrett og privatliv, blant annet gjennom vern om den personlige integritet mv. Skal hindre makt- og myndighetsmisbruk. Viktig for sårbare grupper | Ikke-terapeutisk forskning fordrer frivillig samtykke. Vern om privatliv og den personlige integritet mv. Skjerper legalitetsprinsippet ved at det settes opp et krav om lovhjemmel for enkelte inngrep |
| Alminnelig erstatnings- rett | Forhindre uforsvarlig og skade-voldende handlinger | I utgangspunktet alle og all virksomhet | Innebærer i realiteten et krav om forsvarlig virksomhet | Både subjektivt (uaktsomhets) ansvar og objektivt ansvar er aktuelt. Ansvaret kan følge av så vel ulovfestet som lovfestet rett |
| Pasientskadeloven | Forhindre skader | Offentlig helsetjeneste samt helsepersonell. Gjelder skader voldt bl.a. ved forsøk | Innebærer i realiteten et krav om forsvarlig virksomhet | Tilnærmet objektivt ansvar |
| Produktansvarsloven | Forhindre skader | Erstatningsansvar for skade som voldes av produkt | Aktuell ved utprøving av legemidler. Generell forsikringsplikt, samt spesiell forsikringsplikt | Personskade voldt av legemiddel (legemiddelskade) eller under utprøving av legemiddel (forsøksskade) erstattes |
| Straffeloven | Forhindre uønskede handlinger | Alle | Setter opp forbud mot visse handlinger og gjør overtredelse straffbar | Forskning på mennesker vil ofte innebære et fysisk eller psykisk inngrep i den personlige integritet. Slike inngrep kan anses som en straffbar legemskrenkelse. Samtykke vil kunne gjøre inngrepet straffri |
| Patentloven | Sikre opp- finnerens rettigheter | Forskere og forsknings- institusjoners rettigheter | Åpner for muligheten til å ta patent, dvs. få enerett til kommersiell utnyttelse | |
| Åndsverks-loven | Sikre om opphavs- personens rettigheter | De som produ- serer åndsverk | Et forskningsresultat vil kunne være et åndsverk. Publikasjoner er normalt åndsverk | Den som skaper et åndsverk, feks. et vitenskapelig verk, har opphavsrett til verket. Gir enerett til å råde over åndsverket mv. |
| Arbeidstaker-oppfinnel- sesloven | Regulere forholdet mellom oppfinner og arbeids-giver | Arbeidstakeres oppfinnelser | Sikrer forskere enkelte rettigheter,f.eks. publiseringsrett | Arbeidstakere har i utg.pkt. samme rett til sine oppfinnelser som andre oppfinnere. Varslingsplikt. Arbeidsgiver kan kreve å få overført retten til oppfinnelsen til seg |
| Lov om universiteter og høyskoler | Sikre god utdanning og faglig utvikling (forskning) | Universitetet og høgskoler | Forskningsplikt. Formidlingsplikt. Regler om organisering av universiteter og høgskoler | |
| Helseper-sonelloven | Sikre kvalitet i helse-tjenesten | Helsepersonell som yter helsehjelp – både off. og privat | Berører forskning mer indirekte. Gråsone (grader av overlapp) mellom helsehjelp og forsk-ning, samt mellom helsepersonell og forsker | Mange generelle reguleringer av interesse. Fremstår som en generell ”hovedlov” på helserettens område |
| Pasientrettighets-loven | Sikre be-folkningen god helsehjelp | Pasienter gis rettigheter overfor helse- tjenesten | Gråsone mellom helsehjelp og forskning – pasient og forsøks- person – behandler og forsker | Mange generelle reguleringer av interesse, særlig om medbestemmelsesrett. Fremstår som en generell ”hovedlov” |
| Psykisk helsevern- loven | Sikre forsvarlig psykisk helsevern | Psykisk helsevern | Tvangsmessig terapeu- tisk forsøksvirksomhet er ikke tillatt | Ikke-terapeutisk forskning faller trolig utenfor loven |
| Spesialist- helse- tjeneste- loven | Fremme folkehelsen og helse- tjenestens kvalitet | Spesialist- helsetjenesten | Forskningsplikt for sykehus | Regulerer en rekke forhold. Herunder helsepersonells plikter, samt organisering av spesialisthelsetjenesten |
| Helsetilsyns-loven | Sikre helse-tjenestens kvalitet | Statens helse- tilsyn – Helsetilsynet i fylkene | Statens helsetilsyn skal føre faglig tilsyn med helsetjenesten og helse- personell | Forholdet til forskning er noe uklart. Tilsynet omfatter terapeutisk forskning og forsk-ning foretatt av helsepersonell |
| Transplantasjons- loven | Transplantasjon, sykehusobduksjon, bruk av fostervev og avgivelse av lik | Flere bestemmelser av direkte betydning. Forbud mot transplantasjon i forskningsøyemed. Regulerer bruk av fostervev etc. | Departementet skal godkjenne bruk av fostervev | |
| Bioteknologi-loven | Sikre forsvarlig bruk av biotekno-logi | Humanmedisinsk bruk av bioteknologi. Gjelder i utgangspunktet ikke ikke-tereapeutisk forsk- ning, unntak kap. 3 | Forbud mot forskning på befruktede egg med mer, fremstilling av menneskeembryoer ved kloning med mer, kloning. Genterapi i forskningsøyemed forbudt | Medisinsk bruk av bioteknologi krever godkjenning av departementet |
| Smittevern-loven Med forskrifter | Vern mot smitsomme sykdommer | Åpner for register- forskning | ||
| Gentekno- logiloven | Sikre forsvarlig frem- stilling og bruk av genmodifiserte organismer | Gjelder bl.a. forskning som innebærer fremstilling og bruk av genmodifiserte organismer | Regulerer sammen med biotenknologiloven genterapi | Konkrete forsøk som involverer genterapi, kan unntaksvis godkjennes av Sosial- og helsedirektoratet |
| Biobank- loven | Sikre forsvarlig innsaml., oppbevar., etc. av humant biologisk materiale | Biobanker | Regulerer forsknings- biobanker | Regler om innsamling, opp-bevaring, behandling, og destruksjon av humant bio-logisk materiale. Regler om opprettelse av forskningsbiobanker. Skal godkjennes av REK og departementet |
| Legemiddellloven jf. forskrift om klinisk utprøving av legemidler | Sikre forsvarlig utprøving. Vern av deltakere | Klinisk utprøving av legemidler | Regulerer utprøving av legemidler | Felles regelverk i hele EØS området basert på direktiv 2001/20/EC |
10.2 Lover og forskrifter av særlig interesse
10.2.1 Generelt
I dette punktet skal formålet og det saklige anvendelsesområdet til enkelte lover som kan komme til anvendelse på medisinsk og helsefaglig forskning, kort omtales. For flere av lovenes vedkommende er det saklige virkeområdet noe uklart i forhold til medisinsk og helsefaglig forskning.
10.2.2 Transplantasjonsloven
Formålet med lov om transplantasjon, sykehusobduksjon og avgivelse av lik m.m. (transplantasjonsloven) er å regulere adgangen til å foreta slike inngrep. Det overordnede formål er å sikre etisk forsvarlig virksomhet. Loven er fragmentarisk og regulerer flere forskjellige forhold, og kan komme til anvendelse på medisinsk forskning og uttak av humant biologisk materiale. Loven bestemmer at organer eller annet biologisk materiale kan tas fra person som har gitt skriftlig samtykke, til behandling av sykdom eller legemsskade hos en annen, så sant dette ikke medfører noen nærliggende fare for giverens liv eller helse, jf § 1, 1. ledd. Lovens § 2 regulerer adgangen til å ta organer og annet biologisk materiale fra en avdød. Et nytt kapittel II A regulerer adgangen til å benytte fostervev fra provosertaborterte fostre til visse formål (medisinsk forskning, diagnostikk, fremstilling av vaksine og behandling, jf § 8b, 1. ledd). For medisinsk forsknings vedkommende, sier loven at fostervev bare kan brukes til dette dersom det ikke finnes andre likeverdige metoder, jf. § 8b, 2. ledd. Loven bestemmer at lik på bestemte vilkår kan kreves avgitt til bruk for undervisning og forskning i anatomi og andre medisinske fag ved universitetene, jf. § 9. Lovens § 6 slår forøvrig fast at uttak av blod, fjerning av mindre hudpartier og andre mindre inngrep som kan likestilles med disse, ikke omfattes av bestemmelsene i transplantasjons-loven.
10.2.3 Legemiddelloven med forskrift om klinisk utprøving av legemidler
Lov om legemidler m.v. (legemiddelloven) av 12. april 1992 gjelder legemidler og andre preparater til medisinsk bruk. I § 2 heter det at ”Med legemidler forstås i denne lov stoffer, droger og preparater som er bestemt til eller utgis for å brukes til å forebygge, lege eller lindre sykdom, sykdomssymptomer eller smerter, påvirke fysiologiske funksjoner hos mennesker eller dyr, eller til ved innvortes eller utvortes bruk å påvise sykdom.” I § 3 heter det at ”Kongen gir forskrifter om klinisk utprøving av legemidler på mennesker og dyr. I forskriften kan bl.a. fastsettes hva som skal regnes som klinisk utprøving av legemidler, at slik utprøving skal godkjennes av den myndighet Kongen bestemmer og de nærmere vilkår for godkjenning.” Dette er gjort ved forskrift om klinisk utprøving av legemidler av 24. september 2003.
Forskriften gjelder klinisk utprøving av legemidler på mennesker, og omfatter altså både pasienter og friske personer. Forskriften omfatter ikke utprøvende behandling på enkeltpasienter eller ikke-intervensjonsstudier, (§ 1-1). Med klinisk utprøving forstås enhver systematisk studie av legemidler til mennesker i den hensikt å skaffe til veie eller etterprøve kunnskap om legemidlenes effekter eller påvirkning av fysiologisk funksjon, interaksjoner, bivirkninger, opptak, fordeling, metabolisme og utskillelse, eller for å studere deres terapeutiske verdi, (§ 1-2).
Forskriftens formål er å sikre medisinsk og etisk forsvarlig klinisk utprøving av legemidler, samt internasjonal harmonisering av regelverket ved at forskriften innarbeider EUs legemiddeldirektiv.
10.2.4 Menneskerettsloven
Lovens formål er å styrke menneskerettighetenes stilling i norsk rett (§ 1). Loven innarbeider fire av de mest sentrale menneskerettighetskonvensjonene (§ 2) i norsk rett. Bestemmelsene i konvensjoner og protokoller som er nevnt i § 2 skal ved motstrid gå foran bestemmelser i annen lovgivning (§ 3). Menneskerettighetene kommer til anvendelse på all offentlig virksomhet, herunder forvaltningen, forskning, domstolene og lovgivning.
10.2.5 Pasientrettighetsloven
Lov av 2. juli 1999 nr. 63 om pasientrettigheter trådte i kraft 1. januar 2001, og loven har samlet alle rettigheter pasienter har innenfor helsetjenesten i en felles lov. Loven innebærer i stor grad en videreføring av regler som tidligere var spredt på flere lover, og en kodifisering/formalisering av tidligere ulovfestet rett. Loven innfører imidlertid også en rekke nye regler. Lovens formål er i henhold til § 1-1:
å bidra til å sikre befolkningen lik tilgang på helsehjelp av god kvalitet ved å gi pasienter rettigheter overfor helsetjenesten. Lovens bestemmelser skal bidra til å fremme tillitsforholdet mellom pasient og helsetjeneste og ivareta respekten for den enkelte pasients liv, integritet og menneskeverd.
Med pasient forstås i loven ”en person som henvender seg til helsetjenesten med anmodning om helsehjelp, eller som helsetjenesten gir eller tilbyr helsehjelp i det enkelte tilfelle” (§ 1-3).
Lovens forhold til medisinsk og helsefaglig forskning blir dermed som helsepersonelloven, jf. nedenfor.
10.2.6 Helsepersonelloven
Lov av 2. juli 1999 nr. 64 om godkjenning av helsepersonell m.v. (helsepersonelloven) trådte i kraft 1. januar 2001, og avløser en rekke særlover for de ulike helseprofesjoner.
I lovens § 1 heter det at ”(L)ovens formål er å bidra til sikkerhet for pasienter og kvalitet i helsetjenesten samt tillit til helsepersonell og helsetjeneste.”
Av lovens § 2 første ledd fremgår det at loven gjelder helsepersonell og virksomheter som yter helsehjelp i riket. Begrepet ”helsehjelp” er definert i § 3 tredje ledd hvor det heter: ”Med helsehjelp menes enhver handling som har forebyggende, diagnostisk, behandlende, helsebevarende eller rehabiliterende mål og som utføres av helsepersonell.” Av ordlyden fremgår dermed at loven i utgangspunktet ikke gjelder medisinsk forskning. Indirekte vil imidlertid enkelte bestemmelser kunne tenkes å komme til anvendelse ved medisinsk forskning, for eksempel der forskning er kombinert med behandling. Grensen mellom klinisk virksomhet og medisinsk forskning kan i mange tilfeller være uklar.
10.2.7 Personopplysningsloven
Personopplysningsloven trådte i kraft 1. januar 2001, og erstattet den tidligere lov om personregistre fra 1978. Loven innarbeider EUs personverndirektiv (Direktiv 95/46/EF) i norsk rett.
Av lovens formålsbestemmelse i § 1 første ledd fremgår at loven ”skal beskytte den enkelte mot at personvernet blir krenket gjennom behandling av personopplysninger.” Dette utdypes i andre ledd hvor det fremgår at loven skal bidra til at personopplysninger behandles i samsvar med grunnleggende personvernhensyn, noe som eksemplifiseres ved begreper som ”personlig integritet, privatlivets fred og tilstrekkelig kvalitet på personopplysninger.” I lovens § 2 nr. 1 er personopplysninger definert som ”opplysninger og vurderinger som kan knyttes til en enkeltperson.” En undergruppe av personopplysninger er sensitive personopplysninger, jf. § 2 nr. 8. For slike opplysninger gjelder det strengere regler.
10.2.8 Helseregisterloven
Formålet med helseregisterloven er ”å bidra til å gi helsetjenesten og helseforvaltningen informasjon og kunnskap uten å krenke personvernet, slik at helsehjelp kan gis på en forsvarlig og effektiv måte. Gjennom forskning og statistikk skal loven bidra til informasjon og kunnskap om befolkningens helseforhold, årsaker til nedsatt helse og utvikling av sykdom for administrasjon, kvalitetssikring, planlegging og styring. Loven skal sikre at helseopplysninger blir behandlet i samsvar med grunnleggende personvernhensyn, herunder behovet for personlig integritet, privatlivets fred og tilstrekkelig kvalitet på helseopplysninger” (§ 1).
Loven gjelder for behandling av helseopplysninger i helseforvaltningen og helsetjenesten som skjer helt eller delvis med elektroniske hjelpemidler, og som skjer for å fremme formål som beskrevet i § 1. Loven gjelder også for annen behandling av helseopplysninger i helseforvaltningen og helsetjenesten til slike formål, når helseopplysningene inngår eller skal inngå i et helseregister. Loven gjelder både offentlig og privat virksomhet, jf § 3.
Med helseopplysninger forstås ”taushetsbelagte opplysninger i henhold til helsepersonelloven § 21 og andre opplysninger og vurderinger om helseforhold eller av betydning for helseforhold, som kan knyttes til en enkeltperson” (§ 2). Med behandling av helseopplysninger forstås ”enhver formålsbestemt bruk av helseopplysninger, som f.eks. innsamling, registrering, sammenstilling, lagring og utlevering eller en kombinasjon av slike bruksmåter”, jf. § 2.
10.2.9 Biobankloven
Biobanklovens formål er ”å sikre at innsamling, oppbevaring, behandling og destruksjon av materiale som inngår i en biobank foretas på en etisk forsvarlig måte, og at biobanker utnyttes til individets og samfunnets beste. Dette skal skje i samsvar med grunnleggende personvernhensyn, prinsipper om respekt for menneskeverd, menneskerettigheter og personlig integritet, og uten diskriminering av mennesker som det biologiske materialet stammer fra” (§1, 1. ledd).
Loven skal legge til rette for at materialet i biobanken kan benyttes til helsemessige formål, herunder diagnostikk, behandling, forskning og undervisning, på en etisk forsvarlig måte (§ 1, 2. ledd). Loven gjelder også forskningsbiobanker, samt organiseringen av denne virksomheten. Med mindre annet følger av Biobankloven, skal helse- og personopplysninger som utledes fra humant biologisk materiale, behandles etter personopplysningsloven, helseregisterloven, helsepersonelloven og eventuelt annen lovgivning som særlig regulerer vern av personopplysninger, jf. § 3, 2. ledd. Det innebærer at Biobanklovens bestemmelser har forrang fremfor bestemmelser i de nevnte lover når også disse kommer til anvendelse.
Biologisk materiale som uttas i forbindelse med undersøkelse, diagnostikk og behandling, og som destrueres etter kort tid, omfattes ikke av loven. Loven gjelder likevel dersom materialet blir brukt til forskning, jf. § 3, 1. ledd. Loven definerer ”forskningsbiobank” som en samling humant biologisk materiale og opplysninger som direkte fremkommer ved analyse av dette materialet, og som anvendes eller skal anvendes til forskning” (§2, 2. ledd).
Med humant biologisk materiale forstås i denne lov organer, deler av organer, celler og vev og bestanddeler av slikt materiale fra levende og døde mennesker.” (§ 2, 3. ledd).
10.2.10 Bioteknologiloven
Formålet med Bioteknologiloven er ”å sikre at medisinsk bruk av bioteknologi utnyttes til beste for mennesker i et samfunn der det er plass til alle. Dette skal skje i samsvar med prinsipper om respekt for menneskeverd, menneskelige rettigheter og personlig integritet og uten diskriminering på grunnlag av arveanlegg basert på de etiske normer nedfelt i vår vestlige kulturarv” (§ 1-1).
Loven gjelder humanmedisinsk bruk av bioteknologi m.m. og omfatter assistert befruktning, forskning på befruktede egg og kloning, fosterdiagnostikk, genetiske undersøkelser av fødte og genterapi m.m., jf. § 1-2, 1. ledd. Loven gjelder ikke for forskning som ikke har diagnostiske eller behandlingsmessige konsekvenser for deltakeren eller hvor opplysninger om den enkelte ikke føres tilbake til vedkommende, jf. § 1-2, 2. ledd. Unntatt fra dette er bestemmelsene i lovens kapittel 3, som regulerer adgangen til å forske på befruktede egg, kloning m.m. Loven gjelder heller ikke for obduksjon som faller inn under transplantasjonsloven kapittel II og sakkyndig likundersøkelse, jf. straffeprosessloven § 228.” (§ 1-2, 3. ledd).
Lovens forhold til medisinsk og helsefaglig forskning har vist seg noe vanskelig å håndtere pga ulike tolkninger.
10.3 Profesjonsnormer, ikke-rettslige veiledninger m.m.
10.3.1 Helsinki-deklarasjonen
Helsinki-deklarasjonen 27 er den mest sentrale profesjonsnorm på området for medisinsk forskning i dag – både i Norge og internasjonalt. Helsinki-deklarasjonen ble utarbeidet av Verdens legeforening i 1964, og er senere blitt endret flere ganger. Deklarasjonen er ikke juridisk bindende, men er etter hvert blitt innarbeidet og bredt akseptert ikke bare i profesjonen selv, men også av nasjonale myndigheter, blant annet gjennom anvendelse av de regionale forskningsetiske komiteer. Enkelte av deklarasjonens prinsipper kan derfor trolig anses som alminnelige rettslige prinsipper, som for eksempel prinsippet om informert samtykke.
Deklarasjonen etterfulgte Nürnberg-kodeksen av 1947 og foreningens Genève-erklæring av 1948. 28 Den viktigste forskjellen fra Nurnberg-kodeksen er at Helsinki-deklarasjonen utvides til å omfatte klinisk forskning, altså både terapeutisk og ikke-terapeutisk forskning. Forskning er dermed ikke begrenset til forsøk på friske forsøkspersoner, men kan også omfatte pasienter.
For å forhindre misbruk av individer i forskningens tjeneste og at forskningens nytteverdi bare blir vurdert ut fra hva som er til nytte for fellesskapet, fastholder Helsinki-deklarasjonen at hensynet til den enkelte forsøksperson alltid må gå foran vitenskapens og samfunnets interesser. Deklarasjonen understreker at ansvaret for forsøkspersonen alltid påhviler legen og aldri vedkommende selv, selv om hun eller han har gitt sitt samtykke.
Forskningen må følge ”generelt aksepterte vitenskapelige prinsipper” (I,1) og utføres av ”vitenskapelig kvalifiserte personer” (I,3). Prosjektet må beskrives i en protokoll. Hensikten med forskningen må stå i et ”rimelig forhold til risikoen for forsøkspersonen” (I,4). Det må foretas en ”omhyggelig vurdering av risikomomenter” (I,5). Disse hensynene må ivaretas før man henvender seg til forsøkspersoner. Forsøkspersonen skal få ”fyllestgjørende underretning om formål, metoder, ventede fordeler og mulige farer i forbindelse med undersøkelsen samt om det ubehag som den kan medføre”. Informasjonen skal foreligge skriftlig. Forsøkspersoner skal bli informert om at de fritt kan avstå fra deltakelse, og at de kan trekke seg på et hvilket som helst tidspunkt (I,9). Når forsøkspersonene har fått tid til å vurdere deltakelse, skal de avgi et skriftlig samtykke. Hvis et prosjekt utvikler seg annerledes enn forutsatt, skal forsøkspersonen bli informert og kunne føle seg trygg på at prosjektlederen avslutter prosjektet ”hvis man finner at farene er større enn fordelene” (I,7). Protokoll, risikonytte-vurdering og ivaretakelsen av et frivillig informert samtykke skal fremlegges for og vurderes av en forskningsetisk komité som er uavhengig av de forskerne som ønsker å gjennomføre studien. 29
10.3.2 Vancouver-konvensjonen
Et annet sentralt dokument utarbeidet av legeprofesjonen er Vancouver-konvensjonen 30 av 1988 som er revidert flere ganger senere. Den er utarbeidet av International Committee of Medical Journal Editors. Konvensjonen stiller opp krav som de fleste medisinske tidsskrift bruker ved publisering av vitenskapelige artikler. Konvensjonen gir både praktiske og etiske retningslinjer for forfattere. Blant annet må Helsinki-deklarasjonen være fulgt og forskningsprosjektet må være tilrådd av en uavhengig etisk komité. Konvensjonen blir anvendt av over 500 medisinske tidsskrifter verden over.
10.3.3 CIOMS Internasjonale etiske retningslinjer for biomedisinsk forskning som involverer mennesker
The Council for International Organizations of Medical Sciences (CIOMS) formulerte i 1982 ”Internasjonale etiske retningslinjer for biomedisinsk forskning som involverer mennesker”. 31 Retningslinjene ble revidert i 1993 og sist i 2002.
CIOMS ble etablert i 1949 av Verdens helseorganisasjon (WHO) og UNESCO, men til forskjell fra de to FN-organene er CIOMS en veldedig, ikke-statlig organisasjon (NGO, Non-governmental organization). CIOMS fremstår som en paraply-organisasjon og en rekke sentrale NGOer er medlemmer; blant annet Verdens legeforening. I tillegg er Norges forskningsråd og NEM medlemmer. Det at organisasjonen er en NGO bestående hovedsakelig av bransjens egne organisasjoner, gjør at retningslinjene fremstår som profesjonsnormer. Organisasjonens sterke tilknytning til mellomstatlige og statlige organisasjoner gjør imidlertid at de har en litt annen karakter enn rene profesjonsnormer. Retningslinjene er ikke juridisk bindende i Norge, men må antas å ha en viss tyngde som veiledende normer.
10.3.4 Etiske regler for leger
De etiske regler for leger 32 er utarbeidet av Den norske lægeforening (Legeforeningen), første gang i 1960 og senere revidert flere ganger, senest i 2002. I de etiske reglene heter det blant annet at:
En lege skal verne menneskets helse. Legen skal helbrede, lindre og trøste. Legen skal hjelpe syke til å gjenvinne sin helse og friske til å bevare den.
Legen skal bygge sin gjerning på respekt for grunnleggende menneskerettigheter, og på sannhet og rettferdighet i forhold til pasient og samfunn.
Ved utprøving av nye metoder skal hensynet til forsøkspersonen være det primære.
En lege bør etter sin kompetanse bidra til utvikling og formidling av den medisinske viten.
Reglene retter seg først og fremst mot behandlingssituasjonen, men inneholder også bestemmelser som er relevante for forskning.
Rådet for legeetikk, nedsatt av Legeforeningen, fører tilsyn med at de etiske regler for leger overholdes.
10.3.5 Retningslinjer for samarbeid mellom legestand og farmasøytisk industri
Legeforeningen har i samarbeid med Legemiddelindustriforeningen utarbeidet retningslinjer for samarbeid mellom leger og farmasøytisk industri. 33 Der heter det blant annet at:
Samarbeid mellom legestand og farmasøytisk industri er en forutsetning for utvikling av nye, og forbedring av eksisterende legemidler og behandlingstilbud. Det bør herske et godt, åpent og ryddig forhold mellom farmasøytisk industri og legestand. Begge parter, og befolkningen for øvrig, er tjent med et samarbeid som utvikles uten at det oppstår avhengighet. … Klinisk legemiddelutprøving er en særlig viktig del av den medisinske og farmakoterapeutiske forskning. Virksomheten styrker fagmiljøet i Norge, og er av stor betydning for nasjonal såvel som internasjonal legemiddelutvikling. Leger forvalter gjennom sin legemiddelforskrivning pasientenes interesser såvel som store offentlige ressurser. Det er derfor viktig at samarbeid mellom legestand og farmasøytisk industri ivaretas på en slik måte at det ikke skapes avhengighetsforhold. … All kontakt mellom den farmasøytiske industrien og legestanden skal være redelig og åpen for innsyn og kontroll … Begge parter har ansvar for at internasjonale konvensjoner såvel som gjeldende lover og bestemmelser for kliniske utprøvinger etterleves.
10.3.6 Tannleger
Flere lover og forskrifter regulerer tannlegers yrkesutøvelse. I tillegg har Tannlegeforeningen vedtatt Etiske regler for tannleger, som alle medlemmer er forpliktet til å følge. Verken rettsreglene eller profesjonsnormene retter seg spesielt mot tannlegers forskningsvirksomhet. Det er således noe uklart hvilket regelverk tannleger forholder seg til i sin forskningsvirksomhet.
Den norske tannlegeforenings råd for tannlegeetikk er foreningens høyeste organ i spørsmål som gjelder tannlegeetikk og kollegiale forhold. Det har til oppgave å utrede etiske problemer som det blir forelagt og bidra til at regelverket gjøres kjent for medlemmene. Tannleger som driver klinisk utprøving av legemidler er omfattet av regelverket for dette.
10.3.7 Psykologer
Psykologer er underlagt de samme lover og regler som gjelder for annet helsepersonell. I tillegg har Norsk psykologforening sluttet seg til ”Etiske prinsipper for nordiske psykologer”. Disse prinsippene kommer til anvendelse på all virksomhet som utøves av psykologer, også forskning. Prinsippene er imidlertid generelle og ikke spesielt rettet inn mot forskning. Det finnes i tillegg internasjonale profesjonsnormer, tilsvarende Helsinki-deklarasjonen, som regulerer psykologers forskningsvirksomhet spesielt. I tillegg finnes lokale retningslinjer ved de enkelte psykologiske fakultetene. En gjennomgang av de lokale retningslinjer etterlater et inntrykk av at mye overlates til hver enkelt psykologs skjønn, og at det er behov for et klart rammeverk.
10.3.8 Etiske retningslinjer for sykepleieforskning i Norden
Norsk sykepleierforbund har sluttet seg til Sykepleiernes Samarbeid i Norden (SSN), som består av de nordiske sykepleierforbundene, og ”Etiske retningslinjer for sykepleieforskning i Norden”. Det heter her at ”Sykepleie er uløselig forbundet med respekten for menneskerettigheter, herunder retten til liv og verdighet og til å bli behandlet med respekt”. De vitenskapsetiske krav bygger på de etiske prinsipper som kommer til uttrykk i FNs menneskerettighetserklæring og i Helsinki-deklarasjonen. Prinsippene angir hovedretningslinjer for god etisk standard i forskning som involverer mennesker. Sykepleieforskning veiledes av prinsippet om autonomi, om å gjøre godt, om ikke å forvolde skade og av prinsippet om rettferdighet.
10.3.9 Forskningsetisk sjekkliste
Forskningsetisk sjekkliste 34 er en fempunktsliste utarbeidet av Den nasjonale forskningsetiske komité for naturvitenskap og teknologi (NENT). Listen er uavhengig av fagområde og oppsummerer det som komiteen anser er viktigst å avklare i forbindelse med et forskningsprosjekt. Denne sjekklisten blir også benyttet ved søknader om prosjektmidler fra Norges forskningsråd:
Prosjektets mål og metode
Vil prosjektets mål og metoder bryte med alment aksepterte verdisyn? Dette kan f.eks. gjelde hvis:
prosjektet bidrar til å øke kontrollen med og manipulering av enkeltindivider i samfunnet
miljøhensyn ikke blir tilstrekkelig ivaretatt
prosjektet har diskutable militære/forsvarsmessige implikasjoner
prosjektet bryter med norsk lov ( f.eks. dyrevernloven eller forskrifter om biologiske forsøk)
Forskning der forsøkspersoner er involvert
Blir informert samtykke fra forsøkspersonene innhentet på en forsvarlig måte?
Er det klart at det ikke foreligger avhengighetsforhold som kan tenkes å påvirke forsøkspersonens samtykke?
Persondata
Blir alle persondata tilstrekkelig anonymiserte slik at et adekvat personvern er sikret?
Risiko og sikkerhet
Vil selve gjennomføringen av prosjektet kunne medføre skade på mennesker, dyr eller natur av et omfang som ikke bør neglisjeres? Er i så fall de impliserte personer innforstått med det?
Whistle-blowing - innebygget varslingssystem
Vil en prosjektmedarbeider som får alvorlige forskningsetiske betenkeligheter ved gjennomføringen av et forskningsprosjekt, ha anledning til å legge disse frem for en uavhengig høringsintans? Vil dette være kjent og avklart på forhånd?
10.3.10 Inkludering av kvinner
Den nasjonale forskningsetiske komité for medisin (NEM) ble bedt av Sosial- og helsedepartementet om å følge opp NOU 1999:13 om Kvinners helse i Norge 35 . I denne utredningen pekes det på at det ikke tas tilstrekkelig hensyn til kjønn som variabel i medisinsk forskning. På denne bakgrunn har NEM utarbeidet retningslinjer for inkludering av begge kjønn i medisinsk forskning, med særlig vekt på inkludering av kvinner i fertil alder og gravide, og behovet for kjønnsspesifikke analyser. 36
Disse retningslinjene er bygget inn i skjemaet for vurdering i REK, som også håndhever retningslinjene.
10.3.11 Oppdragsforskning
Oppdragsforskning er et område som særlig utfordrer forskningens uavhengighet. Den nasjonale forskningsetiske komité for samfunnsvitenskap og humaniora (NESH) vedtok en sjekkliste for forskningsetiske hensyn som bør inkluderes i oppdragskontrakter 26. november 1999. Den er senere endret.
NESH understreker at oppdragsforskning utgjør en viktig del av forskningen. Samtidig er det av stor viktighet for denne forskningen at den bevarer sin uavhengighet, både av hensyn til troverdighet og av hensyn til de langsiktige brukerinteressene. Oppdragsforskning utgjør en betydelig del av den samlede forskningen både innen samfunnsvitenskap og humaniora. Det har imidlertid ifølge NESH rådd forholdsvis stor uklarhet om plikter og rettigheter, og om forventninger og krav til relasjonen mellom oppdragsgiver, forskere og de institutter de eventuelt er knyttet til.
NESH legger til grunn at ”oppdragsforskning er kjennetegnet ved at oppdragsgiver og oppdragstaker har inngått en kontrakt om at et forskningsarbeid skal utføres, slik at oppdragsgiveren eller andre interesserte parter bidrar til å finansiere forskningen, til å formulere problemstillinger og har interesse i å bruke resultatene.” For at det skal være snakk om ”oppdragsforskning”, må oppdragsgiver og/eller brukere ha en formell eller faktisk rett til å utøve påvirkning av eller kontroll med prosjektet. Omfanget av betaling for forskningen er ikke avgjørende for om det dreier seg om et oppdrag. Heller ikke er det av betydning at finansieringskilder og brukere er de samme. Den som finansierer forskningen kan for eksempel delegere til en annen part retten til å delta i problemformuleringene. Den institusjonelle rammen, om forskningen formidles via et marked eller gjennom forskningsråd, er heller ikke i seg selv avgjørende. Et generelt poeng i retningslinjene er understrekningen av forskningsinstitusjonenes etiske ansvar, både når det gjelder interne forhold og institusjonenes forhold til parter utenfor.
Sjekklisten er generell og kan anvendes i forbindelse med alle typer oppdragsforskning, ikke bare samfunnsvitenskapelig, for å sikre forskningens frie og sannhetssøkende natur, ved å fremme oppdragsforskningens faglige integritet.
11 Menneskerettighetene
11.1 Oversikt
Det menneskerettslige rammeverk ble etablert etter annen verdenskrig, først og fremst som en direkte følge av de umenneskelige handlinger som ble begått i tyske konsentrasjonsleire. Verden fikk blant annet innblikk i grove overgrep mot fanger som ble utsatt for tvungne medisinske eksperimenter. Å fremme universell respekt for og overholdelse av menneskerettighetene er et av hovedformålene med De forente nasjoner (FN), jf. FN pakten artiklene 1, 55 og 56.
FNs Verdenserklæring om menneskerettigheter av 1948 slår fast prinsippet om at alle mennesker uavhengig av rase, kjønn, religion og politisk tilhørighet er født frie og like med den samme verdighet og rettigheter, og angir en katalog over fundamentale menneskerettigheter. Verdenserklæringen er ikke i seg selv juridisk bindende, men de prinsipper den gir uttrykk for har senere fått folkerettslig status gjennom internasjonale konvensjoner, jf. særlig FN-konvensjonen om sivile og politiske rettigheter og FN-konvensjonen om økonomiske, sosiale og kulturelle rettigheter, begge fra 1966.
Menneskerettighetene omfatter blant annet retten til liv, politisk frihet og personlig sikkerhet, rettferdig rettergang, fravær av tortur og umenneskelig behandling, vern om ytringsfrihet og privatliv, samt individets rett til sosial trygghet, en tilfredsstillende levestandard, utdanning og til den høyest oppnåelige fysiske og psykiske helse. Av særlig interesse i forskningssammenheng er artikkel 7 i konvensjonen om sivile og politiske rettigheter, som oppstiller et krav om informert samtykke ved medisinske og vitenskapelige eksperimenter.
Selv om menneskerettighetene formelt ble introdusert ved opprettelsen av FN i 1945, er det først i løpet av de siste ti årene at de har oppnådd en slik anerkjennelse at de fremstår som sentrale og anerkjente rettsprinsipper med stor utbredelse. Av spesiell interesse for medisinsk og helsefaglig forskning er Europarådets Konvensjon om menneskerettigheter og biomedisin (1997), som i fortalen henviser til Verdenserklæringen om menneskerettigheter, de to FN-konvensjonene fra 1966 og andre internasjonale konvensjoner. Konvensjonen etablerer grunnleggende menneskerettigheter på det biomedisinske område, herunder kravet om individets frie og informerte samtykke, retten til privatliv og regler for beskyttelse av personer med redusert eller manglende samtykkekompetanse.
Menneskerettighetene er en del av folkeretten, og er juridisk bindende for statene i den grad disse har ratifisert (frivillig sluttet seg til) menneskerettighetskonvensjonene. Statene er folkerettslig forpliktet til å respektere og beskytte borgernes menneskerettigheter i overensstemmelse med konvensjonene. En viktig del av statenes forpliktelse ligger i å vedta og implementere nasjonal lovgivning i overensstemmelse med de internasjonale forpliktelser. Statene er forpliktet til, gjennom lovgivning eller andre tiltak, å sikre respekten for menneskerettighetene.
I 1994 fikk den norske Grunnlov en ny bestemmelse – Grl. § 110 c - som slår fast at det påligger statens myndigheter å respektere og sikre menneskerettighetene (1. ledd), og at nærmere bestemmelser om gjennomføring av traktater skal fastsettes ved lov (2. ledd). For norsk retts vedkommende preges forholdet til folkeretten av dualisme, det vil si at folkerettslige forpliktelser som Norge påtar seg ikke automatisk blir en del av norsk rett. Det må en særlig gjennomføringsakt til for at traktater skal bli en del av norsk rett, for eksempel inkorporasjon av traktater ved lovgivning. Norge har inkorporert fire viktige menneskerettighetskonvensjoner med tilleggsprotokoller ved vedtakelse av lov om styrking av menneskerettighetenes stilling i norsk rett av 21. mai 1999 (Menneskerettsloven). Men selv om en konvensjon ikke er innlemmet i norsk rett, følger det likevel av det såkalte presumsjonsprinsipp at norsk rett må antas å være i overensstemmelse med Norges folkerettslige forpliktelser, og at de norske regler så langt det er mulig skal tolkes slik at det ikke oppstår konflikt med Norges folkerettslige forpliktelser, herunder menneskerettighetene.
I Norge spiller menneskerettighetene og tanken om individets grunnleggende rettigheter en helt sentral plass i rettsutviklingen, også på det helserettslige område. I 1999 ble det vedtatt fire nye helselover i Norge: lov om pasientrettigheter, lov om helsepersonell, en ny lov om psykisk helsevern og lov om spesialisthelsetjenesten. Alle disse lovene er viktige fra et menneske- og pasientrettslig perspektiv. Pasientrettighetsloven står likevel i en særstilling. Denne loven setter pasienten i fokus og gir individuelle rettigheter i forbindelse med helsehjelp i langt større grad enn det som var tilfelle tidligere. Lovens formål er å sikre befolkningen lik tilgang på helsehjelp av god kvalitet. Den gir pasienter rettigheter overfor helsetjenesten, fremmer tillitsforholdet mellom pasient og helsetjeneste og skal ivareta respekten for den enkelte pasients liv, integritet og menneskeverd. Pasientrettighetsloven, i lys av menneskerettighetene, innebærer at rettighetstenkningen er gitt forrang fremfor den tradisjonelle paternalisme som har preget helsepersonells holdning til pasientene i helsevesenet. Nå handler det i langt større grad om individenes rett til å bestemme over seg selv i møtet med helsevesenet.
I det følgende gis det først en oversikt over menneskerettsloven, dernest følger en kort redegjørelse for menneskerettigheter som står sentralt i forhold til medisinsk og helsefaglig forskning. Disse rettigheter er for øvrig implementert i ulike deler av gjeldende norsk lovgivning, og vil omtales i mer detalj nedenfor.
11.2 Menneskerettsloven
Lov om styrking av menneskerettighetenes stilling i norsk rett av 21. mai 1999 (menneskerettsloven) inkorporerer fire av de mest sentrale menneskerettighetskonvensjonene i norsk rett. I følge lovens § 2 skal Den europeiske menneskerettskonvensjon med tilleggsprotokoller, FN-konvensjonen om økonomiske, sosiale og kulturelle rettigheter, FN-konvensjonen om sivile og politiske rettigheter med protokoller og FNs barnekonvensjon gjelde som norsk lov så langt de er bindende for Norge. Loven skal bidra til å redusere den usikkerhet som i noen grad har hersket om menneskerettskonvensjoners rettslige stilling, øke kunnskapen om konvensjonene og signalisere menneskerettighetenes viktige plass i norsk retts- og samfunnsliv, jf. også Grunnloven § 110c. Formuleringen ”styrke menneskerettighetenes stilling” er blant annet valgt for å understreke at menneskerettskonvensjoner som ikke blir inkorporert, likevel er å anse som tungtveiende rettskilder. For eksempel vil Europarådets konvensjon om menneskerettigheter og biomedisin (Oviedo-konvensjonen), som Norge er i ferd med å ratifisere, men som ikke er inkorporert i norsk rett, likevel være en sentral rettskilde ved fastleggingen av gjeldende regler for medisinsk og helsefaglig forskning. Menneskerettslovens § 3 fastslår at bestemmelsene i de inkorporerte konvensjoner og protokoller ved motstrid skal gå foran bestemmelser i annen norsk lovgivning. Dette gir de inkorporerte konvensjoner en spesiell rettslig status, og innebærer at norsk lovgivning – herunder en eventuell ny lov om medisinsk og helsefaglig forskning – må være i overensstemmelse med de inkorporerte menneskerettighetskonvensjoner.
11.3 Kravet om samtykke i artikkel 7 i FN-konvensjonen om sivile og politiske rettigheter
Medisinsk forskning vil i prinsippet kunne innebære eller føre til krenkelse av en rekke sivile og politiske rettigheter, som retten til liv, retten til fravær av tortur eller grusom, umenneskelig eller nedverdigende behandling, retten til frihet og sikkerhet, og retten til privatliv. Av særlig interesse er Konvensjonen om sivile og politiske rettigheter artikkel 7, som direkte retter seg mot medisinske eller vitenskapelige eksperimenter:
Ingen må utsettes for tortur eller for grusom, umenneskelig eller nedverdigende behandling eller straff. I særdeleshet må ingen, uten sitt frie samtykke, utsettes for medisinske eller vitenskapelige eksperimenter.
Bestemmelsen innebærer at Nürnberg-kodeksens krav om samtykke til medisinske eksperimenter er slått fast i en internasjonal og juridisk forpliktende sammenheng, som nå også er innarbeidet i norsk lov og har forrang foran andre lover ved eventuell motstrid. Bestemmelsen omfatter ikke behandlende inngrep som skjer utelukkende i pasientens interesse, men tar sikte på medisinsk forsøksvirksomhet der den svake part – forsøkspersonen eller pasienten - i en samhandlingssituasjon vil kunne tjene som middel for forskerens eller samfunnets vitenskapelige interesser. Det primære formål med artikkel 7 er å beskytte forsøkspersoner mot risikofylte eller farlige medisinske eksperimenter.
Kravet om ”fritt samtykke” må forstås som et frivillig, aktivt og positivt tilsagn om å delta i forsøket. 37 Det må ikke foreligge tvang eller utilbørlig påvirkning av forsøkssubjektet, og samtykket må være uttrykkelig. Det betyr at stilltiende samtykke som regel ikke vil være en akseptabel samtykkeform ved medisinsk forskning. 38 Kravet om fritt samtykke er et absolutt krav, og kan ikke fravikes ved lovgivning.
11.4 Retten til privatliv i Den europeiske menneskerettighets-konvensjon artikkel 8 og FN-konvensjonen om sivile og politiske rettigheter artikkel 17
Artikkel 8 i Den europeiske menneskerettighetskonvensjon
Også Den europeiske menneskerettighetskonvensjon er inkorporert i norsk rett ved menneskerettsloven. Konvensjonens artikkel 8 slår fast at
Enhver har rett til respekt for sitt privatliv og familieliv, sitt hjem og sin korrespondanse.
Det skal ikke skje noe inngrep av offentlig myndighet i utøvelsen av denne rettighet unntatt når dette er i samsvar med loven og er nødvendig i et demokratisk samfunn av hensyn til den nasjonale sikkerhet, offentlige trygghet eller landets økonomiske velferd, for å forebygge uorden eller kriminalitet, for å beskytte helse eller moral, eller for å beskytte andres rettigheter og friheter.
Kjernen i artikkel 8 er å beskytte individet mot vilkårlige inngrep fra offentlige myndigheters side, og motsvares av prinsippet om at inngrep i individets privatliv m.v. må ha hjemmel i lov (legalitetsprinsippet). Bestemmelsen verner om den enkeltes rett til hemmelighold av personlige opplysninger, så vel som beskyttelse mot handlinger som truer individets fysiske integritet. Kroppen konstituerer den viktigste grensen mellom individet og omverdenen, og er derfor blant de mest beskyttelsesverdige goder. Personlige opplysninger tilhører også den private sfære som individet har sterke beskyttelsesverdige interesser knyttet til.
Artikkel 17 i FN-konvensjonen om sivile og politiske rettigheter
Artikkel 17 i konvensjonen om sivile og politiske rettigheter fastslår at:
Ingen må utsettes for vilkårlige eller ulovlige inngrep i privat- eller familieliv, hjem eller korrespondanse, eller ulovlige inngrep på ære eller omdømme.
Enhver har rett til lovens beskyttelse mot slike inngrep eller angrep
Også denne bestemmelsen er inkorporert i norsk rett ved menneskerettsloven.
Begge de nevnte bestemmelsene bygger på Verdenserklæringen om menneskerettigheter av 1948 artikkel 12. De har blitt tolket dithen at de rommer ganske omfattende personverngarantier i forbindelse med databehandling av personopplysninger. FNs menneskerettighetskomité har uttalt at artikkel 17 krever at behandling av personopplysninger både innen offentlig og privat virksomhet underlegges rettslig regulering i tråd med grunnleggende prinsipper om personopplysningsvern. 39 Den europeiske menneskerettighetsdomstolen legger en lignende oppfatning til grunn ved tolkningen av Den europeiske menneskerettighetskonvensjon artikkel 8. Domstolen har slått fast at når offentlige myndigheter behandler opplysninger om den enkelte uten deres samtykke eller kunnskap, er dette i utgangspunktet å betrakte som et inngrep i deres rett til respekt for sitt privatliv. Inngrepet må derfor kunne rettferdiggjøres i henhold til artikkel 8 andre ledd. Dette innebærer i praksis at databehandlingen må lovreguleres i overensstemmelse med grunnleggende prinsipper om personopplysningsvern. 40 Det vises respekt for individets privatliv når lovgivningen regulerer adgangen til å benytte personopplysninger til ulike formål.
11.5 Europarådets konvensjon om menneskerettigheter og biomedisin (Oviedo-konvensjonen)
Oviedo-konvensjonen 41 er den første folkerettslige konvensjon som er særskilt innrettet mot å ivareta individets rettigheter i forbindelse med biomedisinsk behandling og forskning. Formålet med konvensjonen er å beskytte alle menneskers verdighet og identitet og - uten forskjellsbehandling - respektere deres integritet, rettigheter og fundamentale friheter ved anvendelse av biologi og medisin (artikkel 1). Det slås også fast at hensynet til individets velferd har forrang fremfor rene samfunnsmessige og vitenskapelige hensyn (artikkel 2). Norge undertegnet konvensjonen i 1997, og forventes å ratifisere den med det første. Utvalgets anbefalinger ligger derfor innenfor de rammer som følger av Oviedo-konvensjonen med tilleggsprotokoller.
Oviedo-konvensjonen omfattes ikke av menneskerettsloven, og har dermed ikke samme status som Den europeiske menneskerettighetskonvensjon og FN-konvensjonene. Det følger imidlertid av det alminnelige presumsjonsprinsipp at norsk rett må forutsettes å være i overensstemmelse med konvensjonen når den først er ratifisert. Menneskerettsloven § 1, som slår fast at lovens formål er å styrke menneskerettighetenes stilling i norsk rett, vil ha selvstendig betydning i denne sammenheng.
Konvensjonen oppstiller krav om fritt og informert samtykke (artikkel 5), og slår fast retten til privatliv, samt retten til å få vite hvilke helseopplysninger som er lagret om en selv (artikkel 10). Konvensjonens kapittel V retter seg særskilt mot forskning, og inneholder bestemmelser om beskyttelse av forsøkspersoner, beskyttelse av personer som ikke har samtykkekompetanse og inneholder bestemmelser om forskning på embryoer (artiklene 16, 17 og 18). Det slås videre fast at menneskekroppen og dens deler ikke som sådan skal gi opphav til økonomisk vinning (artikkel 21).
Europarådet har vedtatt en tilleggsprotokoll til Oviedo-konvensjonen for biomedisinsk forskning. 42 Også denne tilleggsprotokollen vil bli forpliktende for Norge.
Protokollen fremhever også prinsippet om at hensynet til den enkelte går foran hensynet til vitenskapens og samfunnets interesser, og at forskningen bare kan rettferdiggjøres hvis den har potensial til å generere vitenskapelig kunnskap som kan danne grunnlaget for helsefremmende tiltak (artiklene 3, 5 og 6). I tillegg slås det fast at forskning på mennesker bare kan rettferdiggjøres når andre sammenlignbare alternativer ikke finnes, og at forskning ikke må involvere risiko for den enkelte som er uforholdsmessig i forhold til mulige vinninger. Tilleggsprotokollen oppstiller krav til forskningen i situasjoner der deltakeren ikke selv vil få nytte av forskningen (artikkel 8), krever godkjenning av forskningsprosjekter av kompetent organ i henhold til nasjonal lovgivning (artikkel 9) og oppstiller kvalitets- og sikkerhetskrav i overensstemmelse med vitenskapelige kriterier (artikkel 10). Tilleggsprotokollen inneholder detaljerte bestemmelser om informasjon og samtykke, samt bestemmelser om beskyttelse av personer som ikke selv er i stand til å avgi samtykke (Artiklene 16-20).
De enkelte bestemmelser i henholdsvis Oviedo-konvensjonen og tilleggsprotokollen omtales nærmere under drøftingen av tilsvarende regler i norsk rett.
11.6 UNESCOs erklæring om menneskets arveanlegg og menneskerettigheter
Norge sluttet seg 11. november 1997 til UNESCOs erklæring om menneskets arveanlegg og menneskerettigheter. 43 Erklæringen er ikke rettslig bindende, men det ligger en moralsk forpliktelse på stater som har sluttet seg til erklæringen om å følge den opp i form av nasjonale retningslinjer eller nasjonal lovgivning. I innledningen blir det pekt på at forskning på menneskets arveanlegg og resultatene av denne forskningen, åpner for enorme muligheter til å forbedre helsen både for den enkelte og for menneskeheten. Det understrekes imidlertid at denne type forskning til fulle må respektere både menneskets verdighet, frihet og menneskerettigheter, herunder et forbud mot alle former for diskriminering på grunnlag av genetiske særtrekk.
Av artikkel 4 fremgår det at menneskets arveanlegg ”in its natural state” ikke skal gi opphav til økonomisk vinning.
Erklæringen inneholder i artikkel 5 regler som beskytter personer som deltar i genetisk forskning. Det gjelder blant annet regler om samtykke, rett til å velge å bli informert om resultatet av genetisk forskning, og taushetsplikt ved bruk av personopplysninger.
Artikkel 10 inneholder en bestemmelse om at genetisk forskning ikke må stride mot respekten for menneskerettigheter, grunnleggende friheter og menneskets verdighet, både når det gjelder den enkelte og grupper av mennesker. Forskning som strider mot menneskets verdighet, for eksempel reproduktiv kloning av mennesker, skal ikke tillates.
I artikkel 15 oppfordres statene til å etablere rammeverk for fri utøvelse av forskning på menneskets arveanlegg i tråd med deklarasjonens prinsipper, og fremhever betydningen av uavhengige og multidisiplinære etiske komitéer for overvåking av forskningen (artikkel 16).
12 Alminnelig forvaltningsrett
12.1 Oversikt
Den alminnelige forvaltningsrett oppstiller regler som er felles for all forvaltningsvirksomhet eller for store deler av den. Forvaltningsloven og offentlighetsloven er sentrale på forvaltningens område, og suppleres av alminnelige forvaltningsrettslige prinsipper. Den spesielle forvaltningsrett gir spesialregler for forskjellige grener av forvaltningsretten. For eksempel er helseretten en spesialgren av den alminnelige forvaltningsrett. Regler for medisinsk forskning kan sees som en gren av helseretten eller som spesielle forvaltningsrettslige regler på forskningens område.
12.2 Forvaltningsorganer og grunnleggende krav til forvaltningsvirksomhet
Grunnlovens § 3 slår fast at ”Den udøvende Magt er hos Kongen”. Begrepet ”udøvende Magt” betegner de deler av statens funksjoner som ikke er lovgivning eller dømmende virksomhet, og refererer til beslutninger som treffes av andre offentlige myndighetsorganer enn domstolene, og faktiske handlinger som er påkrevd for å iverksette slike beslutninger. Den utøvende makt omfatter en rekke ulike offentlige funksjoner, som gjerne kalles forvaltningsvirksomhet, og som innebærer at det treffes beslutninger på vegne av det offentlige, i kraft av fullmakter gitt i lov og organisatoriske beslutninger. 44 De enkelte forvaltningsorganer, som Regjeringen, departementer, direktorater, statlige tilsyn, fylkeskommuner, kommuner, universiteter, sykehus, skoler etc, må betraktes som redskaper for å ivareta generelle offentlige målsettinger om rettssikkerhet for borgerne, forsvarlig virksomhet, effektivitet ved ressursbruk og beslutningsprosedyrer, samt åpenhet og demokratisk kontroll med offentlig virksomhet. Et overordnet mål for all forvaltningsvirksomhet er at den skal sikre en forsvarlig og hensiktsmessig forvaltning til beste for borgerne. Offentlige institusjoner er således ikke bare redskaper for å nå overordnede målsettinger, men er også gjenstand for allmennhetens kontroll for å sikre at målene innfris og at borgernes rettigheter ivaretas.
12.3 Forvaltningsvirksomhet og legalitetsprinsippet
Det kan skilles mellom intern forvaltningsvirksomhet som gjelder organisering av offentlig virksomhet, behandling av saker som ikke skal munne ut i vedtak av betydning for et bestemt individs rettsstilling og forvaltningsvirksomhet rettet mot borgerne. Eksempler på intern forvaltningsvirksomhet er organisering og ledelse av sykehus, universiteter og skoler, samt forholdet mellom ulike forvaltningsorganer (mellom regjeringen og departementene, mellom de ulike departementer, mellom departement og fylkesmann, mellom Statens helsetilsyn og Helsetilsynet i fylkene osv). 45 Slik virksomhet vil kun indirekte ha betydning for borgernes rettsstilling.
Forvaltningsvirksomhet som direkte griper inn i borgernes rettsstilling omfatter både det offentliges privatrettslige virksomhet (som avtaleinngåelser og utøvelse av eierrådighet) og offentlig myndighetsutøving der forvaltningen opptrer som styringsorgan og treffer ensidige beslutninger (”vedtak”) som ”generelt eller konkret er bestemmende for rettigheter eller plikter til private personer”, jf. forvaltningslovens § 2 første ledd bokstav a. Ikke minst spørsmål knyttet til det offentliges myndighet til å treffe beslutninger som ”gjelder rettigheter eller plikter til en eller flere bestemte personer” (”enkeltvedtak”) står sentralt i forvaltningsretten.
I situasjoner der det offentlige treffer vedtak som medfører innskrenkninger i borgernes frihet, for eksempel ved at individet utsettes for uønskede inngrep i sin fysiske frihet, eller må tåle innskrenkninger i sin handlefrihet, kommer kravet om rettssikkerhet særlig på spissen. Det følger av ulovfestet konstitusjonell sedvanerett - det såkalte legalitetsprinsippet - at offentlig myndighetsutøving som griper inn i borgernes frihet, eiendomsrett og rettssfære krever hjemmel i formell lov. Begrunnelsen for legalitetsprinsippet er den moderne rettsstats forutsetning om individets frihet og at innskrenkninger i denne krever et særlig rettslig grunnlag. Legalitetsprinsippet vil for eksempel kunne stå i veien for at det uten hjemmel i lov oppstilles forbud mot visse typer forskning, og mot at individer uten lovhjemmel eller samtykke pålegges å delta i forskning. I tråd med legalitetsprinsippet er den rettslige hovedregel dermed at individet bare blir rettslig forpliktet av det vedkommende selv inngår bindende avtale om eller samtykker til, med mindre det foreligger lovbestemmelser eller andre gyldige rettsgrunnlag som begrenser individets handle-, avtale- eller valgfrihet. 46
Legalitetsprinsippet setter med andre ord klare grenser for det offentliges handlefrihet. Forvaltningsretten kan dermed sies å ha en dobbelt funksjon: Den legger det rettslige grunnlag for at forvaltningen kan styre samfunnet på en effektiv, forsvarlig og hensiktsmessig måte, og setter samtidig grenser for det offentliges frihet til å velge mål og midler.
12.4 Forvaltningsloven og offentlighetsloven
12.4.1 Forvaltningslovens anvendelse
Forvaltningsloven skal sikre en forsvarlig og hensiktsmessig offentlig forvaltning. Den kommer til anvendelse på den virksomhet som drives av forvaltningsorganer, det vil si et hvert organ for stat eller kommune, jf. § 1. Den omstendighet at universitetene og de fleste sykehus er i offentlig eie, gjør at mange leger og forskere i prinsippet er offentlige tjenestemenn og -kvinner hvis virksomhet omfattes av forvaltningsloven. Forvaltningsloven inneholder bestemmelser om habilitet og alminnelige saksbehandlingsregler om taushetsplikt, veiledningsplikt, partsinnsyn osv som vil være relevante for medisinsk forskningsvirksomhet som drives av et forvaltningsorgan. Ikke minst vil Forvaltningslovens bestemmelser om taushetsplikt være sentrale.
I mandatet for de regionale forskningsetiske komitéer, fastsatt av Utdannings- og forskningsdepartementet i 1989 med endringer senest i 2003, heter det nå at forvaltningslovens kapitler I-III, samt §§ 18-20 om partsinnsyn, gjelder for komitéenes virksomhet. Dette innebærer at bestemmelsene om lovens virkeområde, definisjoner, samt bestemmelsene om habilitet og alminnelige regler om saksbehandlingen (veiledningsplikt, saksbehandlingstid, taushetsplikt) gjelder. Det legges til grunn i mandatet at komiteene ikke skal anses for å fatte enkeltvedtak etter forvaltningsloven og at lovens regler om begrunnelse og klage derfor ikke gjelder. 47
12.4.2 Forvaltningslovens bestemmelser om taushetsplikt
I lovens § 13, 1. ledd heter det at ”enhver” som utfører tjeneste eller arbeid for et forvaltningsorgan, plikter å hindre at andre får adgang eller kjennskap til det han eller hun i forbindelse med tjenesten eller arbeidet får vite om ”noens personlige forhold”, ”tekniske innretninger og fremgangsmåter”, samt ”drifts- eller forretningsforhold som det vil være av konkurransemessig betydning å hemmeligholde av hensyn til den som opplysningen angår”. Bestemmelsene om taushetsplikt er av betydning både av hensyn til pasienters rettssikkerhet der forskere som ledd i sin forskning behandler pasientopplysninger,og av hensyn til forskere som utvikler nye medisinske metoder eller produkter som det er ønskelig å hemmeligholde av konkurransehensyn. Forvaltningsloven har også bestemmelser om begrensninger i taushetsplikten av hensyn til forskning. I henhold til forvaltningsloven § 13 d, 1. ledd kan departementet bestemme at opplysninger kan eller skal utleveres til bruk i forskning når ”det finnes rimelig og ikke medfører uforholdsmessig ulempe for andre interesser”. 48 Det kan knyttes vilkår til slikt vedtak om begrensning i taushetsplikten, jfr. § 13 d, 2. ledd. I forvaltningsloven § 13 e, 1. ledd heter det at enhver som utfører tjeneste eller arbeid i forbindelse med en forskingsoppgave som et forvaltningsorgan har støttet, godkjent eller gitt opplysninger undergitt taushetsplikt til, plikter å hindre at andre får adgang eller kjennskap til opplysninger undergitt taushetsplikt. Dette gjelder opplysninger som forskeren får fra et forvaltningsorgan, opplysninger som i forbindelse med forskningsarbeidet er mottatt fra private under taushetsløfte og opplysninger som gjelder personer som står i et avhengighetsforhold til den instans som har formidlet deres kontakt med forskeren. De taushetsbelagte opplysninger kan bare brukes slik det er nødvendig for forskningsarbeidet og i samsvar med de vilkår som måtte være fastsatt i henhold til § 13, d, 2. ledd.
12.4.3 Forvaltningens veiledningsplikt
Forvaltningsorganer har innenfor sitt saksområde en alminnelig veiledningsplikt, der formålet er å ”gi parter og andre interesserte adgang til å vareta sitt tarv i bestemte saker på best mulig måte”, jf. § 11, 1. ledd. Denne bestemmelsen gjelder nå for de regionale etiske komitéer, jf. ovenfor. Det følger allerede av mandatet til komitéene at deres oppgave er å veilede og gi råd til forskere om forskningsetiske spørsmål, og komitéene kan på eget initiativ ta opp til behandling forskningsetiske problemer. 49 I forvaltningslovens § 11, 2. ledd heter det at forvaltningsorganer som behandler saker med en eller flere private parter, av eget tiltak skal vurdere partenes behov for veiledning. Etter forespørsel eller når sakens art eller partenes forhold gir grunn til det, skal forvaltningsorganet gi veiledning blant annet om gjeldende lover og forskrifter og vanlig praksis på området.
12.4.4 Offentlighetsloven
Offentlighetsloven skal blant annet sikre at borgerne kan få innsyn og dermed mulighet til å føre kontroll med forvaltningen. Loven, som har samme virkeområde som forvaltningsloven, vil også kunne være av interesse ved medisinsk forskning, for eksempel ved spørsmål om innsyn i dokumenter som beskriver forskningsvirksomhet i regi av offentlige institusjoner.
Lovens hovedregel er at ”forvaltningens saksdokumenter er offentlige så langt det ikke er gjort unntak i lov eller i medhold av lov. Enhver kan hos vedkommende forvaltningsorgan kreve å få gjøre seg kjent med det offentlige innholdet av dokumenter i en bestemt sak” (lovens § 2). Det gjelder likevel flere unntak, blant annet for opplysninger som er underlagt lovbestemt taushetsplikt (se lovens §5a).
Det følger nå av mandatet for de regionale etiske komitéer at offentlighetsloven gjelder i sin helhet for komiteenes virksomhet.
13 Helserett
13.1 Oversikt
Det har i de senere år kommet flere nye lover som gir uttrykk for grunnleggende rettsprinsipper på helserettens område. Lovgivningen kodifiserer eller etablerer plikter for helsepersonell og helseinstitusjoner og styrker pasientenes rettigheter på flere områder. Biobankloven gir lovbestemmelser som får direkte anvendelse ved medisinsk og helsefaglig forskning som forutsetter bruk av biobanker, mens bioteknologiloven inneholder bestemmelser som forbyr visse typer forskning.
Sentrale rettsprinsipper som respekt for liv, personvern, selvbestemmelse, menneskeverd, taushets- og informasjonsplikt, forsvarlighet og sikkerhet vil naturlig inngå som grunnleggende forventninger også i forhold til medisinsk og helsefaglig forskning. I det følgende gis det derfor en generell omtale av viktige helserettslige prinsipper slik de kommer til uttrykk i konkrete lovbestemmelser på helserettens område. De aller fleste lovbestemmelsene er utformet med tanke på medisinsk eller helsefaglig behandling av pasienter, og ikke med tanke på medisinsk eller helsefaglig forskning. Mange av bestemmelsene må likevel antas å komme til anvendelse i den grad handlingene utføres av helsepersonell, eventuelt støttet av analogibetraktninger basert på at tilsvarende hensyn gjør seg sterkt gjeldende. 50
13.2 Respekt for og beskyttelse av menneskeverdet
Den gjeldende helserettslige regulering må sies å bygge på respekt for og beskyttelse av menneskeverdet, herunder respekt for menneskelivet og prinsipper om individets integritet og personvern.
I pasientrettighetsloven slås det fast at loven skal ivareta respekten for den enkelte pasients liv, integritet og menneskeverd (§ 1-1). Biobankloven legger til grunn at biobanker skal brukes i samsvar med grunnleggende personvernhensyn, prinsipper om respekt for menneskeverd, menneskerettigheter og personlig integritet, og uten diskriminering av mennesker som det biologiske materialet stammer fra (§ 1). Tilsvarende formuleringer finnes også i bioteknologilovens § 1-1. Selv om disse prinsipper er nedfelt i nokså vage formålsbestemmelser i lovgivningen, må de antas å ha selvstendig rettslig betydning ved tolking av lovens øvrige bestemmelser. Formålsbestemmelsene kan for øvrig sies å gi uttrykk for grunnleggende rettsprinsipper nedfelt i menneskerettighetene, som naturligvis står helt sentralt også ved regulering av medisinsk og helsefaglig forskning.
Norsk rett inneholder også bestemmelser som ut fra de nevnte overordnede rettsprinsipper forbyr visse typer forskning. I henhold til lov om humanmedisinsk bruk av bioteknologi m.m. (bioteknologiloven) § 3-1 gjelder det et forbud mot å forske på befruktede egg, menneskeembryoer og cellelinjer som er dyrket ut fra befruktede egg eller menneskeembryoer. I § 3-2, 1. ledd er det videre nedlagt forbud mot å fremstille menneskeembryoer ved kloning, 51 å forske på cellelinjer som er dyrket ut fra menneskeembryoer ved kloning og å fremstille embryoer ved kloning ved at arvemateriale fra et menneske settes inn i en eggcelle fra dyr. Det gjelder også et forbud mot bruk av teknikker med sikte på å fremstille arvemessig like individer (§ 3-3).
Oviedo-konvensjonen nedlegger forbud mot å fremstille menneskelige embryoer for forskningsformål, men forbyr ikke forskning på humane embryoer. Det oppstilles imidlertid et krav om at lovgivningen gir adekvat beskyttelse av embryoet (Artikkel 18).
Lov om transplantasjon, sykehusobduksjon og avgivelse av lik (transplantasjonsloven) fikk i 2001 et nytt kapittel II A om bruk av fostervev, i loven definert som celler og vev fra provosertaborterte fostre, jf. § 8a. Det heter i § 8b, 1. ledd at slikt fostervev bare kan brukes til medisinsk forskning, diagnostikk, fremstilling av vaksine og behandling, men at det bare er tillatt dersom det ikke finnes andre likeverdige metoder (§ 8b, 2. ledd 2. setning). Av § 8c fremgår det at bruk av fostervev til behandling og forskning skal godkjennes av departementet. Det kan diskuteres om det eksisterende forbud mot å forske på befruktede egg, menneskelige embryoer og cellelinjer i bioteknologilovens kapittel 3 harmonerer med bestemmelsene i transplantasjonslovens kapittel II A, som begrenser, men ikke forbyr, adgangen til å forske på fostervev. Utvalget ønsker å påpeke problemstillingen, men har ikke hatt anledning til å gå nærmere inn på den.
13.3 Kravet om forsvarlighet
Kravet om forsvarlighet er et grunnleggende krav i helseretten så vel som i den alminnelige forvaltningsrett. Det overordnede formål med forsvarlighetskravet er å sikre at de tjenester som ytes er forenlig med kravet om sikkerhet for pasienter og kvalitet i helsetjenesten, samt tillit til helsepersonell og helsetjeneste, jf. helsepersonelloven § 1.
Kravet om forsvarlighet kommer til uttrykk i helsepersonelloven § 4, 1. ledd, der det heter at helsepersonell skal utføre sitt arbeid i samsvar med de krav til faglig forsvarlighet og omsorgsfull hjelp som kan forventes ut fra helsepersonellets kvalifikasjoner, arbeidets karakter og situasjonen for øvrig. I Ot.prp. nr. 13 (1998-99) om lov om helsepersonell m.v. er det under pkt. 4.2.6.3 inntatt enkelte uttalelser om at lovens forsvarlighetskrav vil gjelde i tilknytning til forskning, utprøving, forsøk eller eksperimentell virksomhet:
Forskning og utprøving av nye metoder må i utgangspunktet ikke skje uten at retningslinjer for slik virksomhet følges. Det gjelder bl a krav til innhenting av samtykke, sikkerhet og dokumentasjon. Hvilke risiki som kan tas innenfor forsvarlighetskravet, vil bero på pasientens tilstand og på alternative behandlingsmuligheter. Pasienten skal så vidt mulig høres og gis adekvat informasjon om formålet med og fremgangsmåten ved forsøket, samt om alternativer og risiki. Det er også en forutsetning at man blir gjort oppmerksom på at metoden primært benyttes som ledd i forsøksvirksomhet. Kravet til at pasientens informerte og velforståtte samtykke må innhentes, skjerpes dersom behandlingen bærer preg av forsøk eller eksperimentering. Videre må det stilles strenge krav til vurderingen av om det var forsvarlig å igangsette forsøket, og om den benyttede fremgangsmåte var faglig forsvarlig. Det vil imidlertid være situasjoner hvor ren eksperimentell behandling vil kunne ansees forsvarlig, f eks der slik behandling utgjør siste forsøk på å redde liv.
Forsvarlighetsvurderingen samt vilkårene for gyldig samtykke er noe annerledes ved ren forsøksvirksomhet enn i de tilfeller formålet med å benytte den utradisjonelle metode først og fremst er å oppnå en helbredende eller lindrende effekt hos vedkommende pasient. Men også i sistnevnte tilfelle må det stilles strenge krav.
Det eksisterer altså et skjerpet krav til forsvarlighet ved forsøksvirksomhet og bruk av utradisjonelle metoder. 52
Den virksomhet det her vises til kjennetegnes imidlertid av at den skjer i forbindelse med undersøkelse og behandling av pasienter, altså virksomhet som allerede av den grunn er omfattet av loven.
Det følger av forarbeidene at forsvarlighetskravet er ment å innebære mer enn et krav om aktsomhet. Uaktsom atferd vil med andre ord alltid være uforsvarlig. 53 Et hovedelement i forsvarlighetskravet er at helsepersonell som utgangspunkt ikke skal gå inn i situasjoner de ikke er kvalifisert for å håndtere, samt at de har plikt til å holde seg faglig à jour. Det følger av helsepersonelloven § 16 at virksomhet skal organiseres på en slik måte at helsepersonellet blir i stand til å overholde lovpålagte plikter og av § 17 at helsepersonell av eget tiltak skal gi tilsynsmyndighetene informasjon om forhold som kan medføre fare for pasienters sikkerhet.
13.4 Kravet om samtykke
13.4.1 Innledning
Individets rett til å bestemme over seg selv er en grunnleggende verdi i vårt samfunn som også gjelder også ved pasientbehandling og medisinsk og helsefaglig forskning. I en behandlings- og forskningsrettslig sammenheng kan det være hensiktsmessig å dele den individuelle selvbestemmelsesrett inn i tre dimensjoner:
Retten til å bestemme over egen kropp
Retten til å bestemme over materiale som stammer fra egen kropp.
Retten til å råde over bruken av opplysninger om en selv.
Disse tre dimensjonene i selvbestemmelsesretten kommer særlig til uttrykk gjennom krav om samtykke til henholdsvis
Fysiske og psykososiale inngrep.
Innsamling, bruk og lagring av humant biologisk materiale.
Innsamling, bruk og lagring av helseopplysninger.
I det følgende gis det en oversikt over viktige bestemmelser om informert samtykke i norsk rett, herunder samtykkekrav og unntak fra dette i ulike sammenhenger, og bestemmelser om samtykkekompetanse og informasjonsplikt. Bestemmelsene om samtykke i biobankloven er viet særlig oppmerksomhet, siden disse gjøres til gjenstand for vurdering senere i innstillingen. Fremstillingen suppleres med relevante konvensjonsbestemmelser og profesjonsnormer om samtykke og informasjon ved medisinsk og helsefaglig forskning. Fremstillingen omfatter også bestemmelser om samtykke, samtykkekompetanse og informasjonsplikt i pasientrettighetsloven til tross for at denne loven ikke gjelder for forskning. Det må likevel antas at samtykkebestemmelsene i denne loven vil kunne ha stor overføringsverdi i forhold til medisinsk og helsefaglig forskning, og vil også i enkelte tilfeller komme til anvendelse, for eksempel der forskning foregår i kombinasjon med behandling.
13.4.2 Krav om samtykke og unntak fra dette i norsk lovgivning
13.4.2.1 Pasientrettighetslovens bestemmelser om samtykke og informasjon
Pasientrettighetslovens § 4-1, 1. ledd slår fast hovedregelen om samtykke ved helsehjelp, og lyder slik:
Helsehjelp kan bare gis med pasientens samtykke, med mindre det foreligger lovhjemmel eller annet gyldig rettsgrunnlag for å gi helsehjelp uten samtykke. For at samtykket skal være gyldig, må pasienten ha fått nødvendig informasjon om sin helsetilstand og innholdet i helsehjelpen.
Bestemmelsen må betraktes som en kodifisering av den rettstilstand som forelå allerede før vedtakelsen av pasientrettighetsloven. Kravet om samtykke er et krav om en faktisk og viljemessig tilslutning til den aktuelle helsehjelp. Av pasientrettighetslovens § 4-2 første ledd, følger det at samtykke kan gis både uttrykkelig og stilltiende, og at stilltiende samtykke anses å foreligge dersom det ut fra pasientens handlemåte og omstendighetene for øvrig er sannsynlig at hun eller han godtar helsehjelpen.
Med uttrykkelig samtykke menes et samtykke i ord, enten skriftlig eller muntlig. Sier pasienten ”ja”, men tar visse forbehold, foreligger fremdeles et uttrykkelig samtykke, men et begrenset sådant. Samtykket trenger som hovedregel ikke være skriftlig, med mindre dette følger av særlige lovbestemmelser.
Også et stilltiende samtykke krever at samtykket på en eller annen måte må ha kommet til uttrykk overfor helsepersonellet, men dette kan skje på annen måte enn ved bruk av ord, for eksempel i form av nikking eller at man rekker ut armen som tegn på at man aksepterer at blodprøve tas. Ved stilltiende samtykke, som også betegnes ”konkludent atferd”, må pasientens samlede atferd og omstendighetene for øvrig gi tilkjenne og sannsynliggjøre at hun eller han godtar helsehjelpen.
Såkalt ”hypotetisk samtykke”, som ikke forekommer i loven, men som har vært brukt i juridisk teori, betegner at det ikke foreligger noe samtykke, heller ikke i form av konkludent atferd, men at det likevel legges til grunn at pasienten ville ha samtykket om vedkommende hadde kunnet eller blitt spurt. Denne form for ”samtykke” har liten legitimitet og begrunnelse som selvstendig rettsgrunnlag etter utviklingen av regler om informert samtykke. Helsepersonellovens § 7 om øyeblikkelig hjelp slår fast at helsepersonell straks skal gi den helsehjelp de evner når det må antas at hjelpen er påtrengende nødvendig. Den underliggende betraktning er at det normalt vil foreligge et ”hypotetisk” samtykke fra pasienten til dette. 54 Helsepersonellovens § 7 bestemmer imidlertid at helsepersonells inngrepsplikt, med de begrensninger som følger av pasientrettighetslovens § 4-9, gjelder selv om pasienten motsetter seg helsehjelpen, noe som innebærer at synsvinkelen hypotetisk samtykke ikke lenger er treffende. I situasjoner der pasienten ikke er i stand til å ivareta sine interesser, vil det i en behandlingssammenheng kunne være aktuelt å bygge på den ulovfestede læren om negotiorum gestio (ubedt hjelp). Forutsetningen er da at det ikke nødvendigvis foreligger en situasjon som betinger øyeblikkelig hjelp, men at pasienten likevel har behov for helsehjelp. Samtidig er det en forutsetning at det er umulig å innhente et uttrykkelig samtykke fra pasienten, men at det foreligger en presumsjon for at pasienten ville ha samtykket om vedkommende hadde vært i stand til det. Det må også foreligge en klar interesseovervekt i favør av inngrepet. 55
Pasientrettighetsloven sier at pasientens samtykke kan være uttrykkelig eller stilltiende, men gir utover dette ingen anvisning på hvilken samtykkeform som skal velges i ulike situasjoner. Dette er i seg selv noe oppsiktsvekkende, siden det klart nok ikke er likegyldig hvilken samtykkeform som velges i ulike situasjoner. Et uttrykkelig samtykke gir som hovedregel et bedre holdepunkt for pasientens holdning til inngrepet enn et stilltiende samtykke. 56 Et alminnelig prinsipp går ut på at jo alvorligere og mer risikofylt et inngrep er, jo strengere krav må det stilles til pasientens samtykke. 57 Det fremgår av forarbeidene til pasientrettighetsloven at sidestillingen av uttrykkelig og stilltiende samtykke er tilsiktet, men at helsepersonellet må forsikre seg om at samtykke foreligger i situasjoner preget av risiko, smerte, upåregnelige konsekvenser eller lignende, og at det vil måtte stilles strengere krav til sannsynligheten for at pasienten har samtykket dersom tiltaket er av mer inngripende karakter. 58 Sterke reelle hensyn tilsier at stilltiende samtykke ikke kan godtas i situasjoner der det er snakk om alvorlige tiltak. Med ”alvorlig” tiltak menes tiltak som innebærer skade eller betydelig smerte eller ubehag, eller risiko for slike virkninger. I slike situasjoner må det kreves et uttrykkelig samtykke fra pasienten. 59
Pasientens samtykke må være ”informert” for å være gyldig, det vil si at den samtykkende må ha en viss innsikt i og forståelse av det samtykket gjelder. Pasientrettighetslovens § 3-2, 1. ledd slår fast at pasienten ”skal ha den informasjon som er nødvendig for å få innsikt i sin helsetilstand og innholdet i helsehjelpen. Pasienten skal også informeres om mulige risikoer og bivirkninger”. I lovens § 3-5 heter det om informasjonens form at den ”skal være tilpasset mottakerens individuelle forutsetninger, som alder, modenhet, erfaring og kultur- og språkbakgrunn” (1. ledd), og at helsepersonellet så langt som mulig skal ”sikre seg at pasienten har forstått innholdet og betydningen av opplysningene” (2. ledd). I forarbeidene til pasientrettighetsloven sies det at informasjonen etter § 3-2 må være tilstrekkelig til at pasienten kan benytte sin rett til medvirkning. Det forutsetter et krav om samtykke og innebærer at pasienten ikke bare har rett til å avstå fra uønsket helsehjelp, men også har rett til selv ”å medvirke ved valg mellom tilgjengelige og forsvarlige undersøkelses- og behandlingsmetoder”, jf. pasientrettighetslovens § 3-1, 1. ledd. 60
Både lovtekst, forarbeider og reelle hensyn tilsier at det er den individuelle pasients behov for informasjon som er avgjørende for informasjonspliktens innhold. Pasienten har krav på tilstrekkelig informasjon om sin helsetilstand, om helsehjelpens innhold og formål og om risiko for skade. Pasienten har krav på å få all den informasjon som er nødvendig for å sette vedkommende i stand til å treffe en selvstendig beslutning med hensyn til om helsehjelpen skal iverksettes eller ikke. Sentralt står informasjon om risiko for utilsiktede skadevirkninger. Rettspraksis tilsier at det selv i situasjoner med minimal risiko foreligger informasjonsplikt, så sant skadefølgen kan bli alvorlig for den enkelte pasient. 61
Det følger av pasientrettighetslovens § 4-1, 2. ledd at pasienten kan trekke sitt samtykke tilbake. Trekker pasienten samtykket tilbake, skal den som yter helsehjelp gi nødvendig informasjon om betydningen av at helsehjelpen ikke gis.
13.4.2.2 Pasientrettighetslovens bestemmelser om samtykkekompetanse
Pasientrettighetsloven inneholder også bestemmelser om hvem som har samtykkekompetanse. Hovedregelen er at myndige personer, samt mindreårige etter fylte 16 år, har samtykkekompetanse, med mindre annet følger av særlige lovbestemmelser, jf. § 4-3, 1. ledd. I forhold til mindreårige etter fylte 16 år er det også et forbehold om at ”tiltakets art” – for eksempel svært alvorlige inngrep - kan tilsi at vedkommende likevel ikke har samtykkekompetanse alene.
Samtykkekompetansen kan falle bort ”helt eller delvis dersom pasienten på grunn av fysiske eller psykiske forstyrrelser, senil demens eller psykisk utviklingshemming åpenbart ikke er i stand til å forstå hva samtykket omfatter”. Et viktig poeng i loven er at samtykkekompetansen bare faller bort så langt det er nødvendig, og at pasientens kompetanse må vurderes konkret. Pasienten kan ha kompetanse til å treffe noen beslutninger, men ikke andre, for eksempel mer sammensatte og vanskelige beslutninger. Det skal en del til for at samtykkekompetansen kan anses for bortfalt, jf. ordet ”åpenbart”. Lettere alderdomssvekkelser eller lettere forstandssvekkelser kan ikke være tilstrekkelig. Loven stiller også et krav om at årsaken til bortfall av samtykkekompetanse må skyldes de i loven nevnte forhold.
Pasientrettighetsloven har også bestemmelser om at foreldre eller andre med foreldreansvar har rett til å samtykke til helsehjelp på vegne av barn, det vil her si personer under 16 år (§4-4, 1. ledd), og likeledes på vegne av ungdom mellom 16 og 18 år som ikke har samtykkekompetanse (§ 4-5, 1. ledd). Helsehjelp til pasienter mellom 16 og 18 år kan imidlertid ikke gis dersom pasienten motsetter seg dette, med mindre annet følger av særlige lovbestemmelser, for eksempel av helsepersonellovens § 7 om øyeblikkelig hjelp.
Pasientrettighetslovens § 4-6 inneholder bestemmelser om samtykke på vegne av myndige personer som ikke har samtykkekompetanse. Hovedregelen er at den som yter helsehjelp kan ta avgjørelse om helsehjelp som er av lite inngripende karakter (1. ledd), mens pasientens nærmeste pårørende kan samtykke til helsehjelp som ikke kan sies å være av lite inngripende karakter (2. ledd). Det er imidlertid et vilkår at helsehjelpen må antas å være i pasientens interesse, og det må være sannsynlig at pasienten ville ha gitt tillatelse til den aktuelle helsehjelp. For å ta stilling til dette, kan det innhentes informasjon fra pasientens pårørende. Det presiseres at helsehjelp etter 1. og 2. ledd ikke kan gis dersom pasienten motsetter seg dette, med mindre annet følger av særlige lovbestemmelser (3. ledd).
Dersom pasienten ikke har nærmeste pårørende, kan den som yter helsehjelp, i samråd med annet kvalifisert helsepersonell, samtykke til helsehjelp for pasienter som mangler samtykkekompetanse (§ 4-8).
Når det gjelder pasienter som er umyndiggjort etter lov av 28. november 1898, skal disse i så stor utstrekning som mulig selv samtykke til helsehjelp. Dersom dette ikke er mulig, kan vergen samtykke på vegne av den umyndiggjorte (§4-7).
Pasientrettighetslovens §§ 4-4 til 4-8 om samtykke på vegne av andre gjelder tilsvarende for personer uten samtykkekompetanse (jf. pasientrettighetslovens § 4-3) som samtykker til innhenting, oppbevaring og behandling av humant biologisk materiale i forbindelse med diagnostikk og behandling, jf. biobanklovens § 11.
13.4.2.3 Biobanklovens samtykkebestemmelser for forskningsbiobanker
Biobankloven gjelder innsamling, oppbevaring og bruk av humant biologisk materiale med tilhørende personopplysninger. Biobanklovens § 12 første ledd gjelder samtykke for forskningsbiobanker, og lyder slik:
Med mindre det foreligger særskilt lovhjemmel eller annet gyldig rettsgrunnlag, krever innsamling, oppbevaring og behandling av humant biologisk materiale til forskningsformål et frivillig, uttrykkelig og informert samtykke fra giveren. Det samme gjelder oppbevaring og bruk av opplysninger knyttet til det biologiske materialet.
I forarbeidene uttales det at det ligger i kravet om uttrykkelig samtykke ”at det ikke er tilstrekkelig med et ”passivt” samtykke. For at et samtykke skal være uttrykkelig må giveren aktivt samtykke til innsamling, oppbevaring og behandling av materialet”. 62 ”Passivt” samtykke betegner det forhold at giveren av et materiale eller opplysninger ikke aktivt protesterer mot en nærmere beskrevet bruk og lagring av materiale eller opplysninger, forutsatt at man er blitt oppfordret til dette hvis man ønsker å reservere seg.
”Passivt” samtykke er ingen etablert samtykkeform i norsk rett, og kan selvfølgelig ikke erstatte et aktivt, uttrykkelig og informert samtykke til å delta i et forskningsprosjekt. ”Passivt” samtykke har først og fremst vært diskutert i forskningssammenheng i forbindelse med ny bruk av et tidligere innsamlet materiale, som deltakeren tidligere har gitt samtykke til bruk av for forskningsformål (se nedenfor).
Det følger av biobanklovens § 12, 2. ledd at samtykket skal kunne dokumenteres og være basert på informasjon om ”formål, metode, risiko, ubehag, konsekvenser og annet av betydning for samtykkets gyldighet.” Kravene til informasjon og spesifikasjon må avgjøres etter en vurdering av risikofaktorer, materialets sensitivitet, forsøkspersonenes sårbarhet og lignende (2. ledd). Kravet om at samtykket skal kunne dokumenteres, indikerer at det senere skal være mulig å fastslå om samtykke er gitt og hva det omfatter. ”Metode” refererer til den metode som brukes for å samle inn og behandle det biologiske materialet. ”Annet av betydning” for samtykkets gyldighet kan for eksempel være retten til å trekke samtykket tilbake og få materialet destruert. Omfanget av informasjonsplikten vil særlig avhenge av forskningens inngripende karakter og opplysningenes sensitivitet. 63
Om kravet til samtykke ved ulike typer forskning uttales det i forarbeidene:
Et samtykke til epidemiologisk forskning og grunnforskning bør for eksempel kunne være relativt overordnet. Epidemiologiske studier og grunnforskning basert på materiale i biobanker innebærer ofte liten eller ingen helserisiko for deltakerne. Det må uansett være en forutsetning at personvernet og sikkerheten blir ivaretatt på en god måte.
Selv om det må være mulig å avgi et samtykke på noe mer overordnet nivå, for eksempel at materialet kan brukes til kreftforskning, hjerteforskning etc., bør generelle utsagn som ”all medisinsk forskning” og ”genetisk forskning” i hovedsak unngås.
Det er en forutsetning at det fremgår eksplisitt av samtykket dersom det biologiske materialet og opplysningene som er utledet ved analyser av materialet, skal benyttes til kommersielle formål.
Samtykke til epidemiologisk forskning og grunnforskning må bl.a bygge på informasjon om mulige fremtidige koblinger til helseregistre og andre helse- og personopplysninger, mulig fremtidig produktutvikling, samarbeid med industrien eller andre former for kommersialisering og næringsutvikling, og muligheter for at materialet kan sendes til utlandet. Dette vil normalt også gjelde for andre typer medisinsk forskning, for eksempel legemiddelforskning, utvikling av diagnostika mv. Det bør også gis informasjon om hvor lenge det innsamlede materialet skal oppbevares”. 64
Biobanklovens ordlyd og forarbeider må sies å legge til grunn et ganske nyansert samtykke-, informasjons- og spesifikasjonskrav som forutsetter en konkret vurdering av forskningens karakter og mulige risikofaktorer, samt andre forhold som vil kunne ha betydning for giveren av materialet. Det sies klart i forarbeidene at samtykke til epidemiologisk forskning og grunnforskning bør kunne være ”relativt overordnet”, forutsatt at det er liten eller ingen helserisiko for deltakerne, samt at personvernet og sikkerheten blir ivaretatt på en god måte. Samtidig presiseres det at samtykket ikke må være altfor generelt, og at deltakeren må få informasjon om kobling til andre registre og opplysninger, utnyttelse til kommersielle formål, samarbeid med industri og andre former for kommersiell utnytting, om materialet skal sendes til utlandet, samt oppbevaringstidens lengde, før samtykke avgis.
13.4.2.4 Unntak fra biobanklovens samtykkekrav
Et generelt unntak fra biobanklovens samtykkekrav følger av passusen ”Med mindre det foreligger særskilt lovhjemmel eller annet gyldig rettsgrunnlag” i lovens 12, 1. ledd. Eksempel på et lovhjemlet unntak er helseregisterlovens § 8, 3. ledd, som bestemmer at Dødsårsaksregisteret, Kreftregisteret, Medisinsk fødselsregister, Meldesystemet for infeksjonssykdommer, Det sentrale tuberkuloseregisteret og System for vaksinasjonskontroll (SYSVAK) kan behandle navn, fødselsnummer og andre direkte personidentifiserende kjennetegn uten samtykke fra den registrerte, i den utstrekning det er nødvendig for å nå formålet med registeret.
I biobanklovens § 12, 4. ledd bestemmes det at transplantasjonslovens regler om presumert samtykke til obduksjon, gjelder tilsvarende for uttak av humant biologisk materiale fra en avdød. Dette betyr at det kan tas ut materiale fra en person som er død på sykehus, sykestuer, fødestuer og sykehjem eller av den som er brakt død til slik institusjon (transplantasjonsloven § 7, 1. ledd) uten uttrykkelig samtykke. Dette gjelder så sant avdøde eller hans/hennes nærmeste ikke har uttalt seg mot det, eller det ikke er grunn til å anta at et slikt inngrep vil være i strid med avdødes eller de nærmestes livssyn, og det heller ikke foreligger andre særlige grunner som taler mot det (jf. transplantasjonsloven § 7, 2. ledd). Uttak av biologisk materiale fra en avdød kan likevel ikke skje før avdødes nærmeste er blitt underrettet om dødsfallet, og det er gått åtte timer etter dødstidspunktet. Foreligger samtykke fra avdøde eller hans/hennes nærmeste, kan uttak av materiale imidlertid skje tidligere (jf. § 7, 3. ledd). Bare dersom det av særlige grunner er nødvendig å få brakt dødsårsaken på det rene uten opphold, kan uttak av materiale foretas uten hensyn til de nevnte vilkår (jf. transplantasjonsloven § 7, 4. ledd).
13.4.2.5 Biobanklovens krav om samtykke ved endret, utvidet eller ny bruk
Biobankloven § 13 første ledd bestemmer at ved endret, utvidet eller ny bruk av tidligere innsamlet materiale og opplysninger som ikke omfattes av det opprinnelige samtykket, skal nytt frivillig, uttrykkelig og informert samtykke innhentes. Dette kan for eksempel være aktuelt dersom materiale som opprinnelig er innsamlet ut fra diagnostiske eller behandlingsrelaterte årsaker, ønskes brukt til forskning. Da skjer det en endret eller ny bruk av materialet som etter loven krever nytt frivillig, uttrykkelig og informert samtykke. Loven skiller ikke mellom små og store endringer, men gjelder etter sin ordlyd enhver endring av bruken av materialet. I og med at forarbeidene til biobankloven klart presiserer hva som menes med et uttrykkelig samtykke, og uttrykkelig utelukker såkalt ”passivt” samtykke, er det nokså klart at denne samtykkeformen vil være i strid med biobanklovens forutsetninger, også ved endret, utvidet eller ny bruk av tidligere innsamlet materiale. 65
Biobankloven er dermed strengere enn Retningslinjene for biomedisinsk forskning som involverer mennesker utarbeidet av The Council for International Organizations of Medical Sciences ( CIOMS ), som i Guideline 6 inneholder en bestemmelse om at det påhviler forskeren å
renew the informed consent of each subject if there are significant changes in the conditions or procedures of the research or if new information becomes available that could affect the willingness of subjects to continue to participate; and, renew the informed consent of each subject in long-term studies at pre-determined intervals, even if there are no changes in the design of objectives of the research.
Disse retningslinjene innebærer at det kun er betydelige endringer (”significant changes”) i forskningsprosjektets prosedyrer eller innhold som krever nytt samtykke fra giveren av materialet. Det kreves også nytt samtykke hvis ny informasjon som kan ha betydning for deltakernes villighet til å delta i prosjektet, blir tilgjengelig. I tillegg kreves det nytt samtykke i langtidsstudier på forhåndsbestemte tidspunkter, selv om det ikke er endringer i forskningen.
Biobanklovens krav om nytt samtykke gjelder likevel ikke dersom det tidligere innsamlede materialet er anonymisert, jf. § 13, 4. ledd. Begrepet ”anonymisert” må forstås i overensstemmelse med helseregisterlovens definisjon av anonyme opplysninger i § 2 nr 3: ”opplysninger der navn, fødselsnummer og andre personentydige kjennetegn er fjernet, slik at opplysningene ikke lenger kan knyttes til en enkeltperson”. Selv om nytt samtykke ikke kreves, må likevel den endrede, utvidede eller nye bruken vurderes av regional komité for medisinsk forskningsetikk.
Det gjelder videre unntak fra kravet om samtykke ved endret, utvidet eller ny bruk dersom ”det er umulig eller svært vanskelig å innhente nytt samtykke”, jf lovens § 13, 2. ledd første setning. I slike tilfeller kan departementet gjøre unntak fra kravet om at det innhentes nytt samtykke. Det må imidlertid foreligge en vurdering fra regional komité for medisinsk forskningsetikk, jf § 13, 2. ledd andre setning. Denne bestemmelsen er særlig aktuell i sammenheng med diagnostiske biobanker og behandlingsbiobanker som inneholder historisk materiale, hvor det ofte vil være umulig eller svært vanskelig å innhente nytt samtykke fra giveren dersom materialet skal brukes i forskning. Bestemmelsen gjelder også for historisk materiale som er innhentet uten samtykke, eller hvor samtykkets innhold og omfang ikke er dokumentert. 66
Unntak fra samtykkekravet til endret, utvidet eller ny bruk gjelder også i situasjoner der giveren av det biologiske materialet er død (§ 13, 3. ledd):
Ved bruk av materiale og opplysninger fra en avdød, skal avdødes antatte vilje og materialets sensitivitet legges til grunn for departementets vurdering. Det skal tas tilbørlig hensyn til avdødes familie og slekt.
Denne bestemmelsen omhandler situasjoner der giveren av det biologiske materialet er død etter avgivelse av det opprinnelige samtykke, men før det er aktuelt med endret, utvidet eller ny bruk av det innsamlede materialet, og derfor ikke kan avgi noe nytt samtykke. Hensynet til avdødes familie og slekt må ivaretas av de regionale forskningsetiske komitéer, og av departementet som har myndighet til å gjøre unntak fra hovedregelen om samtykke, jf. § 13, 2. ledd. Om begrunnelsen for bestemmelsen heter det i forarbeidene at det kan være uheldig at pårørende skal kontaktes og må sette seg inn i og ta stilling til ulike prosjekter på vegne av en avdød slektning, blant annet fordi det kan dreie seg om materiale som er innhentet flere tiår tilbake. 67
13.4.2.6 Biobanklovens bestemmelser om tilbakekall av samtykke, destruksjon m.m.
Biobanklovens § 14 slår fast at den som har avgitt samtykke etter §§ 11-13, til enhver tid kan tilbakekalle slikt samtykke (1. ledd). Det heter videre at dersom samtykket tilbakekalles, kan den som har avgitt samtykke kreve at det biologiske materialet blir destruert. Likedan kan giveren av materialet i en forskningsbiobank kreve at helse- og personopplysninger som er innsamlet sammen med materialet eller som er fremkommet ved analyse eller undersøkelse av materialet, slettes eller utleveres (2. ledd). Disse bestemmelsene er forutsatt harmonisert med retten til å få slettet journalopplysninger etter helsepersonelloven og pasientrettighetsloven, slik at det gjelder samme regler som for journalopplysninger. 68
Det gjelder flere unntak fra retten til å tilbakekalle samtykke eller kreve destruksjon av materiale, sletting eller utlevering av opplysninger etter § 14, 1. og 2. ledd: Retten gjelder ikke dersom materialet er anonymisert, dersom materialet etter bearbeidelse inngår i et annet biologisk produkt, eller dersom opplysningene allerede har inngått i vitenskapelige arbeider. Adgangen til destruksjon gjelder heller ikke dersom det ved lov er fastsatt at materialet eller opplysningene skal oppbevares (§ 14, 3. ledd).
13.4.2.7 Personopplysningslovens og helseregisterlovens samtykkebestemmelser
De samtykkebestemmelser som finnes i personopplysnings- og helseregisterlovgivningen inngår i det som gjerne omtales som personopplysningsvernet, det vil si den enkeltes vern mot at personopplysninger kommer på avveie og eventuelt misbrukes av utenforstående. Helseopplysninger regnes som sensitive personopplysninger i henhold til personopplysningsloven § 2 nr 8.
Det følger av helseregisterlovens § 5, 3. ledd, at før helseopplysninger innhentes for behandling ”skal samtykke fra den registrerte foreligge, hvis ikke annet er bestemt i eller i medhold av lov”. Grunnen er at helseopplysninger er taushetsbelagte opplysninger.
Personopplysningsloven § 2 nr. 7 definerer samtykke som ”en frivillig, uttrykkelig og informert erklæring fra den registrerte om at han eller hun godtar behandling av opplysninger om seg selv”. En tilsvarende definisjon finner vi i helseregisterloven § 2 nr 11. Denne definisjonen samsvarer med biobanklovens krav til samtykke.
Før det overhodet er aktuelt å innhente et slikt individuelt samtykke, må det gis tillatelse til slik behandling i henhold til personopplysningslovens §§ 9 og 33, eller det må følge av lov, og behandlingen må heller ikke være forbudt ved annet særskilt rettsgrunnlag, jf. helseregisterlovens § 5, 1. ledd.
Personopplysninger kan bare behandles dersom ett av vilkårene nevnt i personopplysningslovens § 8 er oppfylt: Oppfyllelse av rettslig forpliktelse, ivareta den registrertes vitale interesser, utføre oppgaver av allmenn interesse m.m. Forskning må betraktes som oppgaver av allmenn interesse. I tillegg må ett eller flere av vilkårene som nevnt i § 9 være oppfylt, for eksempel at den registrerte samtykker i behandlingen eller behandlingen er nødvendig for historiske, statistiske eller vitenskapelige formål, og ”samfunnets interesse i at behandlingen finner sted klart overstiger ulempene den kan medføre for den enkelte”, jf. § 9, 1. ledd bokstavene a og h.
Av personopplysningslovens § 11, 1. ledd bokstav c kan det utledes at det må innhentes nytt samtykke dersom det blir aktuelt å benytte opplysningene til ”formål som er uforenlig med det opprinnelige formålet med innsamlingen”. Tilsvarende bestemmelse finner vi i helseregisterloven § 11, siste ledd. I personopplysningslovens § 11, siste ledd heter det imidlertid at
Senere behandling av personopplysningene for historiske, statistiske eller vitenskapelige formål anses ikke uforenlig med de opprinnelige formålene med innsamlingen av opplysningene, jf. første ledd bokstav c, dersom samfunnets interesse i at behandlingen finner sted, klart overstiger ulempene den kan medføre for den enkelte.
Dette innebærer at når personopplysningene skal brukes til forskning, trenger man ikke innhente nytt samtykke så sant samfunnets interesse i forskningen klart overstiger ulempene den kan medføre for den enkelte. Dette er en sterkt skjønnspreget bestemmelse som åpner for unntak fra samtykkekravet i langt større omfang enn biobankloven, som krever at det må være umulig eller svært vanskelig å innhente nytt samtykke ved endret, utvidet eller ny bruk av materialet for at kravet om innehenting av nytt samtykke skal kunne fravikes, jf biobankloven § 13, 2. ledd. Det følger av biobankloven § 3, 2. ledd (”med mindre annet følger av denne loven…”) at biobanklovens bestemmelser har forrang i situasjoner der også personopplysningsloven kommer til anvendelse. Biobankloven har med andre ord gjort det vanskeligere å endre, utvide eller omdefinere bruken av tidligere innsamlet materiale, herunder opplysninger, siden man må innhente nytt samtykke så sant dette ikke er umulig eller svært vanskelig. Unntak gjelder hvis det dreier seg om anonymisert materiale; da kreves det kun vurdering fra regional komité for medisinsk forskningsetikk (biobankloven § 13, 2. ledd).
13.4.2.8 Samtykke ved klinisk utprøving av legemidler
Forskrift om klinisk utprøving av legemidler til mennesker, gitt med hjemmel i legemiddelloven § 3, inneholder detaljerte bestemmelser om samtykke og informasjon, og representerer et viktig vern om forsøkspersoners fysiske og psykiske integritet og mulighet til å motsette seg å delta i slik utprøvning. I § 4-1 slås det fast at forsøkspersonens informerte samtykke til å delta i utprøvingen skal innhentes før studierelaterte prosedyrer settes i gang. Samtykke skal innhentes etter at utprøver, eller den person som har ledet informasjonsprosessen på dennes vegne, på forhånd har gitt muntlig og skriftlig informasjon om utprøvningen. Det skal foreligge skriftlig bekreftelse fra den som har ledet informasjonsprosessen om at informasjon er gitt. Det følger av bestemmelsen at samtykkeformularet skal inneholde informasjon om blant annet hvem som står bak utprøvningen, hvem som er ansvarlig for å gjennomføre utprøvningen, utprøvningens formål og utførelse, mulig risiko og ubehag, eventuelle alternativer til behandlingen i forsøket, forsikring av forsøkspersoner, kontaktpunkt for å få mer informasjon, og at samtykket når som helst kan trekkes tilbake. Forsøkspersonen skal bekrefte at informert samtykke er gitt, og samtykket skal fortrinnsvis foreligge skriftlig. Muntlig samtykke skal være attestert av uavhengig vitne (§ 4-1, siste ledd).
Forskriften inneholder bestemmelser om klinisk utprøving på personer under 18 år og på personer uten eller med redusert samtykkekompetanse (§§ 4-2 og 4-3). Blant annet heter det i § 4-3 at utprøvning på visse vilkår kan foretas på umyndiggjort eller på myndig person som ikke er i stand til å samtykke på grunn av mangelfulle mentale evner, sykdom eller andre årsaker, etter informert samtykke fra hans eller hennes lovlige representant. Det er et åpent spørsmål hvem som kan regnes som slik lovlig representant.
Av § 4-4 følger det at avgitt samtykke til å delta i en klinisk utprøvning når som helst kan trekkes tilbake. Ved et slikt tilbakekall vil deltakelse i utprøvningen straks opphøre. Pasientdata innsamlet frem til samtykket tilbakekalles vil inngå som studiedata, men ingen ytterligere data skal samles inn etter at samtykket er tilbakekalt.
13.4.2.9 Samtykkekrav ved bruk av fostervev
Av transplantasjonsloven §§ 8b og 8c fremgår det at fostervev kan benyttes i medisinsk forskning så sant det ikke finnes andre likeverdige metoder og dersom departementet har godkjent bruken. Det er et krav at det foreligger et skriftlig samtykke fra kvinnen ”før fostervev kan avgis til en fostervevsbank” (§ 8d, 1. ledd). Først etter at kvinnen har truffet beslutning om svangerskapsavbrudd kan hun stilles overfor spørsmålet om bruk av fostervevet til forskningsformål. Hun må da informeres om den aktuelle bruk, og samtykke ”kan bare gyldig avgis etter at slik informasjon er gitt” (§ 8d, 2. ledd). Disse samtykkebestemmelsene verner om kvinnens rett til å motsette seg uønsket bruk av fostervev som stammer fra hennes kropp. Hun kan motsette seg både at fostervevet brukes til behandling av en annen person og at det brukes til forskning.
13.4.3 Særlige unntak fra samtykkekravet i smittevernloven
Smittevernlovens § 3-7, 1. ledd legger til grunn at tilgjengelig blod, serum og annet biologisk materiale fra mennesker, ikke kan analyseres med henblikk på en smittsom sykdom for et ikke-diagnostisk formål uten samtykke fra de personer som prøvene stammer fra. Med andre ord: Analyser som ikke har et diagnostisk formål, krever samtykke.
I annet ledd oppstilles et unntak fra hovedregelen om samtykke ved ikke-diagnostiske analyser: Laboratorier/institusjoner kan likevel utføre ”kartleggingsundersøkelser” ved bruk av tilgjengelig prøvemateriale som nevnt i første ledd, uten samtykke fra de som har avgitt prøvene. Dette gjelder dersom formålet med undersøkelsen er å overvåke utviklingen av en epidemi som sprer seg i befolkningen, eller belyse forekomsten av en smittsom sykdom i befolkningen eller en del av den. Videre for å bedømme om og i tilfelle hvor godt befolkningen er beskyttet mot en smittsom sykdom som det vaksineres mot, og undersøkelsens resultat er av betydning for smittevernarbeidet. De undersøkelser som tillates gjennomført uten samtykke, må dermed ha en solid smittevernfaglig begrunnelse.
Videre bestemmes det i smittevernlovens § 3-7, 3. ledd at laboratorier/institusjoner også kan utføre ”metodeutprøving” ved bruk av tilgjengelig prøvemateriale som nevnt i første ledd, uten samtykke fra de som har avgitt prøvene, dersom formålet med utprøvingen er å utvikle ny metodikk eller forbedre eksisterende metodikk for påvisning og karakterisering av en smittsom sykdom.
13.4.4 Samtykkekrav i Oviedo-konvensjonen, tilleggsprotokoll for biomedisinsk forskning og internasjonale profesjonsnormer
Anvendelsesområdet for Oviedo-konvensjonen er noe vagt presisert til å gjelde for all anvendelse av biologi og medisin (”with regard to the application of biology and medicine”). I Artikkel 4 heter det at ”[a]ny intervention in the health field, including research, must be carried out in accordance with relevant professional obligations and standards”, og den inkluderer dermed forskning. Konvensjonen oppstiller et krav om fritt og informert samtykke ved enhver helserelatert intervensjon, inkludert forskning, jf. artikkel 5. Formuleringen ”intervention” indikerer at konvensjonen kun omfatter forskning som forutsetter fysiske inngrep, herunder uttak av humant biologisk materiale fra det menneskelige legeme.
I tilleggsprotokoll for biomedisinsk forskning, heter det i artikkel 17: ”No research on a person may be carried out under the provisions of this Chapter without the informed, free, express, specific and documented consent of the person”. Dette tilsvarer bestemmelsen i biobanklovens § 12, bortsett fra at det i sistnevnte ikke står at samtykket skal være spesifisert. Sett i lys av forarbeidene, samt formuleringen om at kravene til informasjon og spesifikasjon må avgjøres etter en vurdering av risikofaktorer, materialets sensitivitet, forsøkspersonenes sårbarhet og lignende, er det imidlertid klart at det gjelder et spesifikasjonskrav etter biobankloven. Kravene til spesifikasjon vil imidlertid kunne variere avhengig av de forhold som er nevnt i loven. Det må dermed kunne konkluderes med at biobankloven tilfredsstiller de samtykkekrav som oppstilles i utkast til tilleggsprotokoll for biomedisinsk forskning artikkel 17.
I artikkel 16 i utkast til tilleggsprotokoll for biomedisinsk forskninger det tatt inn bestemmelser om den informasjonsplikt som påhviler forskeren. Det skal blant annet informeres om ”the nature, extent and duration of the procedures involved, in particular, details of any burden imposed”, ”the risks involved”, og om ”arrangements for access to information relevant to the participant arising from the research and to its overall results”. Det ser ut til at denne bestemmelsen går noe lenger enn biobanklovens § 12, 2. ledd med hensyn til omfanget av informasjonsplikten. Biobankloven sier ingenting om at det skal informeres om mulighetene for å få tilgang til relevant informasjon om den aktuelle forskning og dens resultater. Omfang og varighet er heller ikke uttrykkelig omtalt i biobankloven, men kan innfortolkes (jf. formuleringen ”annet av betydning for samtykkets gyldighet”).
The Council for International Organizations of Medical Sciences ( CIOMS ), etablert i samarbeid mellom WHO og UNESCO i 1949, har utarbeidet internasjonale retningslinjer for biomedisinsk forskning som involverer mennesker (2002). Retningslinjene, som må anses som profesjonsnormer og ikke bindende rettsnormer, inneholder i femte retningslinje omfattende bestemmelser om informert samtykke. Det skal blant annet gis informasjon på en måte som er forståelig for deltakeren, om formålet med forskningen, metoder og forskningsdesign, risikofaktorer, smerte og ubehag, og det skal gis en forklaring på hvordan den aktuelle forskning avviker fra rutinemessig medisinsk behandling. I tillegg oppstilles krav om å informere om varigheten av den enkeltes deltakelse, mulige direkte fordeler for deltakerne av å delta i forskningen, om deltakernes rettigheter, om mulige alternative behandlingsformer, om forskningens sponsorer og institusjonell tilhørighet og annet. Disse retningslinjene oppstiller krav som klart går utover det som kan kreves etter biobankloven.
En annen profesjonsnorm av interesse er Helsinki-deklarasjonen, som i artikkel 22 slår fast at ved ”forskning på mennesker må forsøkspersonen gis fyllestgjørende informasjon om formål, metoder, finansieringskilder, interessekonflikter, forskerens institusjonstilhørighet, forventede fordeler og mulige risikoer i forbindelse med studien, og det ubehag som den kan medføre. Forsøkspersonen skal gjøres kjent med sin rett til ikke å delta i studien, og til på et hvilket som helst tidspunkt å trekke tilbake et gitt samtykke uten frykt for negative konsekvenser. Etter å ha forvisset seg om at forsøkspersonen har forstått informasjonen, skal legen sørge for å få hans eller hennes fritt avgitte informerte samtykke, fortrinnsvis skriftlig. Dersom det ikke er mulig å få samtykket skriftlig, må det ikke-skriftlige samtykket dokumenteres og bevitnes”. I artikkel 23 heter det at legen skal være spesielt varsom hvis forsøkspersonen står i et avhengighetsforhold til ham eller henne eller vil kunne føle seg presset til å gi samtykke. Også Helsinki-deklarasjonens bestemmelser går dermed utover det som kan utledes av biobankloven. Helsinki-deklarasjonens anvendelsesområde er angitt i artikkel 1: ”forskning som omfatter mennesker”, herunder ”forskning på identifiserbart humant materiale eller identifiserbare data”, og har dermed et noe snevrere anvendelsesområde enn biobankloven, som også kommer til anvendelse på anonymisert biologisk materiale.
13.4.5 Om samtykke i kliniske nødsituasjoner
Norsk rett inneholder ingen bestemmelser om hvilke krav til samtykke som gjelder forskning i kliniske nødsituasjoner der det er umulig å innhente samtykke fra personen selv eller en lovlig representant. I tilleggsprotokollen for biomedisinsk forskning 69 artikkel 21 er det gitt bestemmelser om forskning i kliniske nødsituasjoner:
If the research is of a nature such that it can only be undertaken in emergency situations, and where a person is not in a state to give consent and it is impossible to obtain the authorisation referred to in Article 18 paragraph 1.iv, [autorisasjon fra lovlig representant] the law shall determine whether, and under which conditions, this research can take place.
Det finnes også en bestemmelse i Helsinki-deklarasjonen artikkel 26 om forskning i kliniske nødsituasjoner:
Forskning på individer som det ikke er mulig å innhente samtykke fra, heller ikke stedfortredende samtykke eller forhåndssamtykke, kan utføres bare dersom den fysiske eller mentale tilstanden som er til hinder for at informert samtykke kan innhentes, er en nødvendig egenskap ved de personer som inngår i forskningsprosjektet. De særlige grunner som tilsier inklusjon av forsøkspersoner som ikke kan gi eget informert samtykke, skal oppgis i forsøksprotokollen slik at de kan vurderes og godkjennes av den etiske komiteen. Det skal opplyses i protokollen at samtykke til fortsatt deltakelse i forsøket skal innhentes så snart som mulig fra vedkommende selv eller fra person med kompetanse til å handle på vegne av forsøkspersonen på det området det gjelder.
Denne bestemmelsen åpner for forsøk på personer som ikke selv kan samtykke, og der det heller ikke er mulig å innhente samtykke fra en rettslig representant. Pasientens tilstand må være grunnen til at informert samtykke ikke kan innhentes fra denne. Siden det heller ikke er mulig å innhente samtykke fra en representant, synes denne bestemmelsen å være mest aktuell i akutt-situasjoner, der personen er bevisstløs og det ikke er tid til å innhente samtykke fra vedkommendes nærmeste. Dette kan være aktuelt ved et akutt hjerte- eller hjerneinfarkt der pasienten er bevisstløs ved sykehusinnleggelsen, og der vedkommende innlemmes i et forskningsprosjekt om behandling av hjerte- eller hjerneinfarkt. Ved forskning i forbindelse med behandling, kommer også Helsinki-deklarasjonen artikkel 28 til anvendelse, der det heter at medisinsk forskning ”kan kombineres med behandling bare i den utstrekning forskningen berettiges av dens mulige forebyggende, diagnostiske eller terapeutiske verdi”.
13.4.6 Om beskyttelse av personer som ikke har samtykkekompetanse
Biobanklovens § 12, siste ledd bestemmer at for personer uten samtykkekompetanse etter pasientrettighetslovens § 4-3 gjelder pasientrettighetslovens §§ 4-4, 4-5, 4-7 og 4-8 om samtykke til helsehjelp på andres vegne tilsvarende for samtykke til innsamling, oppbevaring og behandling av biobankmateriale til forskning. Disse bestemmelsene er omtalt ovenfor i kapittel 13. Utover dette inneholder norsk rett ingen bestemmelser om beskyttelse av personer som ikke har samtykkekompetanse.
Tilleggsprotokoll til Oviedo-konvensjonen inneholder i kapittel V bestemmelser om dette. Det fremgår av artikkel 18 at forskning på personer som ikke har samtykkekompetanse kan skje dersom samtlige av følgende vilkår er oppfylt:
Hvis resultatene av forskningen vil kunne medføre en reell og direkte fordel for hans eller hennes helse,
Hvis det ikke er mulig å iverksette forskning av sammenlignbar betydning (”effectiveness”) på personer med samtykkekompetanse,
Hvor, hvis mulig, forsøkspersonene har blitt informert om sine rettigheter og om lovreglene som beskytter dem,
Den nødvendige autorisasjon er gitt spesifikt og i skriftlig form av den rettslige representant eller en myndighet, person eller et organ etablert ved nasjonal lovgivning, og etter å ha mottatt den nødvendige informasjon etter Artikkel 19, under hensyntaken til tidligere uttrykte ønsker eller protester. En myndig person uten samtykkekompetanse skal så langt som mulig ta del i autorisasjonsprosedyren. Det skal tas hensyn til oppfatningen til en mindreårig i den grad alder og modenhet tilsier det,
Personen ikke protesterer mot å delta i forskningen.
Artikkel 19 presiserer at de som blir bedt om å autorisere deltakelse av en person i et forskningsprosjekt, skal bli gitt adekvat informasjon i en dokumentert og forståelig form om formål, plan og metoder, samt vurderingen til den etiske komité.
I situasjoner der forskningen ikke vil kunne medføre en reell og direkte fordel for den aktuelle persons helse, kan forskning likevel finne sted dersom vilkårene nevnt under 2, 3, 4 og 5 ovenfor er oppfylt. Dette forutsetter at forskningens målsetting er å bidra, gjennom betydelig forbedring av den vitenskapelige forståelse av personens tilstand, sykdom eller forstyrrelse, til den endelige oppnåelse av resultater som kan være til nytte for den aktuelle person eller for andre personer i samme aldersgruppe, med samme sykdom eller forstyrrelse eller som har samme tilstand. I tillegg må forskningen kun medføre en minimal risiko og en minimal byrde for den aktuelle person. Enhver vurdering av mulige nyttevirkninger av forskningen utover dette, skal ikke kunne brukes til å rettferdiggjøre større risiko eller byrde for personen (artikkel 18 nr. 2).
I artikkel 20 er det presisert at forskning skal anses for å innebære en minimal risiko hvis det kan forventes at den bare vil kunne medføre en veldig liten og midlertidig negativ effekt på helsen til den aktuelle person. Forskningen skal likeledes anses for å innebære en minimal byrde hvis det kan forventes at symptomer eller ubehag kun vil være av veldig lett og midlertidig karakter.
13.5 Taushetsplikt
De bestemmelser om taushetsplikt som følger av forvaltningsloven, er fremstilt ovenfor i kapittel 12. Her skal det kort redegjøres for helselovgivningens bestemmelser om taushetsplikt. Hovedregelen om taushetsplikt for helsepersonell står i helsepersonellovens § 21:
Helsepersonell skal hindre at andre får adgang eller kjennskap til opplysninger om folks legems- eller sykdomsforhold eller andre personlige forhold som de får vite om i egenskap av å være helsepersonell.
Det heter i lovens § 22 at taushetsplikt etter § 21 ikke er til hinder for at opplysninger gjøres kjent for den opplysningene direkte gjelder, eller for andre i den utstrekning den som har krav på taushet, samtykker.
I pasientrettighetslovens § 3-6 første ledd heter det tilsvarende at opplysninger om legems- og sykdomsforhold samt andre personlige opplysninger, skal behandles i samsvar med gjeldende bestemmelser om taushetsplikt. Det heter videre at opplysningene skal behandles med varsomhet og respekt for integriteten til den opplysningene gjelder, og at taushetsplikten faller bort i den utstrekning den som har krav på taushet, samtykker.
Samtykke fra den opplysningene gjelder, er dermed som hovedregel en nødvendig forutsetning for at det kan gjøres unntak fra taushetsplikten. Det finnes imidlertid flere unntak fra dette. Helsepersonellovens § 23 bestemmer at taushetsplikt etter § 21 ikke er til hinder for at opplysninger gis videre til den som før er kjent med opplysningene, når ingen berettiget interesse tilsier hemmelighold, når behovet for beskyttelse må anses ivaretatt ved at individualiserende kjennetegn er utelatt, når tungtveiende private eller offentlige interesser gjør det rettmessig å gi opplysningene videre eller at opplysningene gis videre etter regler fastsatt i lov eller i medhold av lov, når det er uttrykkelig fastsatt eller klart forutsatt at taushetsplikt ikke skal gjelde.
I forbindelse med medisinsk og helsefaglig forskning er det særlig alternativene om at individualiserende kjennetegn er utelatt og at opplysninger kan gis videre i lovbestemte tilfelle, som er aktuelle. Det finnes også en bestemmelse i helsepersonellovens § 29 som sier at departementet kan bestemme at opplysninger kan eller skal gis til bruk i forskning, og at det kan skje uten hinder av taushetsplikt etter § 21. Det presiseres at det kan knyttes vilkår til et slikt vedtak, og at reglene om taushetsplikt etter helsepersonelloven gjelder tilsvarende for den som mottar opplysningene.
Biobanklovens § 16 slår fast at helsepersonellovens bestemmelser om taushetsplikt gjelder tilsvarende for alle som oppretter, oppbevarer, bruker eller på andre måter forvalter eller arbeider ved en biobank. Denne harmoniseringen av regelsettene innebærer en viktig forenkling.
Legemiddellovens § 30 bestemmer at ”[e]nhver som i medhold av stilling eller verv etter loven får kjennskap til drifts- eller forretningshemmelighet, skal med de begrensninger som følger av hans gjøremål etter loven, tie med det han slik får vite. Ingen må i ervervsvirksomhet gjøre bruk av det han slik får vite. Taushetsplikten gjelder også etter at vedkommende har sluttet i stillingen eller vervet. Departementet kan samtykke i unntak fra taushetsplikt etter denne bestemmelse når sterke samfunnsmessige hensyn taler for at opplysningene gis. Taushetsplikten skal likevel ikke være til hinder for utveksling av informasjon (samordning) som forutsatt i lov om Oppgaveregisteret. Forvaltningslovens §§ 13-13e gjelder ikke.”
14 Personvern- og personopplysningsrett
14.1 Oversikt
En viktig del av dagens regulering som gjelder for medisinsk og helsefaglig forskning skal ivareta personvern, og spesielt den delen av personvernet som gjelder forsvarlig behandling av personopplysninger – ”personopplysningsvernet”. Personopplysningsretten inneholder både generelle regler for behandling av personopplysninger (helseregisterloven, personopplysningsloven), samt særskilte regler for opprettelse og bruk av bestemte personregistre, herunder registre i helsesektoren (Dødsårsaksregisteret, Kreftregisteret, Medisinsk fødselsregister mv). I det følgende vil det bli lagt størst vekt på de generelle reglene. Enkelte felles spørsmål vedrørende særskilte registre vil også bli behandlet.
EUs personverndirektiv (95/46/EF) er tatt inn i EØS-avtalen, og Norge er derfor forpliktet til å gjennomføre direktivet på EØS-rettens område. Personopplysningsloven gjennomfører direktivet fullt ut, og er gitt generell anvendelse for all behandling av personopplysninger, uten hensyn til omfanget av EØS-avtalen. I den grad EØS-avtalen tillater det, kan det gis særlovgivning som avviker fra direktivets bestemmelser. I Ot.prp. nr. 5 (1999 – 2000), ved vedtakelsen av helseregisterloven, la regjeringen til grunn at personverndirektivet gjelder for det området denne loven regulerer, noe som innebærer at direktivet også er bindende ramme for behandling av personopplysninger som gjelder helseforhold. Direktivet gjelder således også som ramme for den behandling av helseopplysninger som skjer i tilknytning til helseforskning. Også særlovgivning og -forskrifter vedrørende spesielle helseregistre må være i overensstemmelse med direktivet.
Medisinsk og helsefaglig forskning kan enten være regulert av de alminnelige bestemmelsene i personopplysningsloven, eller komme inn under særreglene i helseregisterloven. Selv om de to lovene har mange likhetspunkter, er det også viktige forskjeller, for eksempel vedrørende krav til rettslig grunnlag for å behandle helseopplysninger. I det følgende vil utvalget primært gjennomgå særreguleringen i helseregiserloven.
Helseregisterloven representerer en særregulering av ”helseopplysninger”, dvs av en viss type personopplysninger som etter personopplysningsloven § 2 nr 8 bokstav c er å anse som sensitive. Personopplysningsloven innebærer regulering av helseopplysninger og andre personopplysninger generelt . Helseregisterloven gjelder databehandling av helseopplysninger når behandlingen skjer innen en viss sektor og for visse formål som er fastsatt i loven. Således gjelder helseregisterloven (litt unøyaktig uttrykt) for databehandling av helseopplysninger i helseforvaltningen og helsetjenesten, når formålet med databehandlingen er å gi effektiv og forsvarlig helsehjelp, eller gi kunnskap i tilknytning til visse administrative og forskningsmessige gjøremål, jf helseregisterloven § 3 (saklig virkeområde) og § 1 (formål). Dersom helseopplysninger i en helseinstitusjon blir behandlet for andre formål enn de som er angitt i helseregisterloven § 1, reguleres ikke forholdet av helseregisterloven, men av personopplysningsloven. Det sentrale poenget er derfor at loven gjelder for bestemte behandlingsformål innen helseforvaltningen og helsetjenesten .
Uttrykket ”helseforvaltningen og helsetjenesten” omfatter alle de institusjoner som har oppgaver i henhold til helselovgivningen, 70 det være seg helsehjelp, planlegging, administrasjon, forskning mv. Uttrykket omfatter både offentlige som private aktører og aktører på alle myndighetsnivåer (stat, fylke, kommune). Databehandling av helseopplysninger utenfor helseforvaltningen og helsetjenesten, skal utelukkende følge bestemmelsene i personopplysningsloven; for eksempel vil sosial- og trygdekontorenes behandling av helseopplysninger følge personopplysningslovens bestemmelser. Avgrensingen til helsetjenesten og helseforvaltningen betyr imidlertid ikke at behandling av helseopplysninger som skjer i institusjoner utenfor denne sektoren, alltid faller utenfor helseregisterloven. Loven gjelder således også i tilfeller der databehandling av helseopplysninger blir satt bort til en databehandler utenfor helsesektoren. Dersom en institusjon som driver helseforskning for eksempel gir analyseoppdrag til en oppdragstaker utenfor helsesektoren, gjelder helseregisterloven således også for denne databehandleren.
Personopplysningsloven spiller en viktig rolle også for behandling av helseopplysninger som hører inn under helseregisterloven (helseregisterloven). Således bestemmer helseregisterloven § 36 at personopplysningsloven med forskrifter gjelder som utfyllende bestemmelser. Hvilke bestemmelser som må regnes som utfyllende, er imidlertid ikke angitt i loven. Meget langt på vei følger helseregisterloven den begrepsbruk og den systematikk som personopplysningsloven anvender. Enkelte av helseregisterlovens bestemmelser er imidlertid basert på et uferdig forslag til personopplysningslov. Avvikene kan være forvirrende, men har neppe reell betydning for hva som må anses å være korrekt fortolkning av loven.
En del av helseregisterloven gjelder adgangen til å samle inn helseopplysninger, opprette helseregistre mv, se lovens kapittel 2. Resten av loven gjelder spørsmål som er parallelle til sentrale deler av personopplysningsloven, dvs alminnelige bestemmelser om hvordan helseopplysninger skal behandles. Innen denne andre delen er det primært sammenhenger mellom de to lovene når det gjelder:
Legaldefinisjoner.
Alminnelige bestemmelser om behandling av opplysninger.
Innsynsrettigheter og informasjonsplikter.
Retting og sletting av opplysninger.
Tilsyn, kontroll og sanksjoner.
Til helseregisterloven er det gitt forskrifter om åtte bestemte særregistre innen helsesektoren som er nærmere omtalt nedenfor. Loven inneholder flere forskriftshjemler der det ikke er gitt særlige forskrifter. I slike tilfelle gjelder personopplysningsforskriften så langt hjemlene i helseregisterforskriften rekker. Av særlig betydning er at hjemlene i helseregisterloven §§ 16 og 17, som innebærer at personopplysningsforskriftens kapittel II om informasjonssikkerhet og kapittel III om internkontroll også får anvendelse på helseregistre og behandling av helseopplysninger. Helseregisterloven har ingen særskilte bestemmelser om konsesjon, men viser i § 5 første ledd til bestemmelsene i personopplysningsloven § 33, noe som også innebærer at deler av at kap. VII i personopplysningsforskriften kommer til anvendelse. Det er egne forskriftshjemler for meldeplikt i helseregisterloven §§ 29 og 30, men særskilte bestemmelser er ikke gitt, noe som innebærer at også bestemmelsene om meldeplikt i personopplysningsforskriftens kap. VII kommer til anvendelse på helseregisterlovens område. Andre deler av samme forskrift kan tenkes å være relevant i forhold til helseregistre, selv om helseregisterloven selv ikke regulerer området. Således vil bestemmelsene i personopplysningsloven og de tilhørende forskriftsbestemmelsene om fjernsynsovervåking og overføring av helseopplysninger til utlandet også gjelde helseregistre og behandling av helseopplysninger ellers.
Det meste av personopplysningsforskriften får altså anvendelse på helseregisterlovens område. Måten denne forskriften kommer inn på avhenger imidlertid av hvorvidt
helseregisterloven regulerer spørsmål i personopplysningslov og / eller –forskrift, og om
helseregisterloven inneholder selvstendige forskriftshjemler eller ikke.
Forholdet mellom de to lovene med tilhørende forskrifter utgjør en komplisert struktur og bør neppe danne mønster for integrering mellom regelverk.
14.2 Personverninteresser
I forarbeidene til helseregisterloven er personvernhensyn tillagt stor vekt:
Helseopplysninger er sensitive opplysninger. Dette tilsier en streng håndtering av rammeverket rundt opplysningene. I de tilfeller det er konflikt mellom hensynet til personvernet til den enkelte pasient eller registrert og forskningens interesser, skal hensynet til personvernet til den enkelte pasient eller registrert i utgangspunktet veie tyngst. 71
Det heter videre at ”grunnleggende personvernhensyn” må forstås på samme måte som i forslag til lov om behandling av personopplysninger. I forarbeidene til personopplysningsloven heter det om dette at det skal legges vekt på:
festnede rettsoppfatninger om personvern, slik disse blir nedfelt i praksis fra domstoler, rettsteori og administrative avgjørelser. 72
Når det gjelder rettspraksis, forelå det i januar 2004 ingen relevante avgjørelser der domstolene har tatt stilling til tolkningen av helseregisterloven, personopplysningsloven og personopplysningsforskriften. En forklaring på dette er at reguleringen er forholdsvis ny. Imidlertid har rettspraksis innen personopplysningsretten tidligere i liten grad dreiet seg om tolking av enkeltbestemmelser, og har i relativt stor grad etablert normer for ulovfestet personvern.
Forarbeidenes henvisning til administrative avgjørelser gjelder primært enkeltvedtak fattet av Datatilsynet og Personvernnemnda. Særlig er det grunn til å vektlegge Personvernnemndas avgjørelse av klagesaker. Det finnes ingen tilgjengelig oversikt over Datatilsynets avgjørelser. Alle saker som avgjøres av Personvernnemnda blir fortløpende publisert på nemndas hjemmeside. 73 I januar 2004 forelå det tre avgjørelser i Personvernnemnda som gjaldt behandling av personopplysninger i forbindelse med medisinsk og helsefaglig forskning.
Når forarbeidene viser til rettsteori, gjelder dette i stor grad en fremstilling av ”personverninteresser” som ble etablert i samband med utarbeidelsen av personregisterloven i 1970-årene, og som senere har vært supplert på bakgrunn av den teknologiske og samfunnsmessige utviklingen. I forarbeidene til helseregisterloven (Ot.prp. nr. 5 (1999 – 2000) er det referert til to ulike fremstillinger av denne teorien. Interesseteori har spilt en stor rolle i tilknytning til arbeidet med alle de sentrale lovene på ”personopplysningsområdet”, og er i tillegg av klar betydning når forvaltningsmyndigheter skal foreta avveininger i konkrete konsesjonssaker mv. Utvalget har derfor funnet det hensiktsmessig å gi en oversikt over det som kan sies å være hovedelementene i personverninteressene. Fremstillingen er gjort kortfattet, fordi disse interessene er beskrevet i tidligere utredninger og for øvrig er gjort utfyllende rede for i faglitteraturen. Utvalget har valgt en fremstillingsmåte som kan sies å kombinere de to fremstillingene av personvern som ble gitt i NOU 1993:22 Pseudonyme helseregistre 74 , samtidig som den på flere punkter er mer detaljert enn tradisjonelle fremstillinger.
Den følgende presentasjonen av personverninteressene er bygget opp rundt fem hovedelementer som typisk uttrykker hvilke interesser enkeltindivider kan ha når andre behandler personopplysninger om dem. Til hver av interessene er det formulert ”krav” som kan sies å øke sannsynligheten for at interessene blir ivaretatt. Interesser og krav er i stor grad basert på generelle erfaringer fra rettssystemet, og innebærer ikke noe postulat om at alle personer har de ulike interessene. Tvert i mot forutsetter interesseteorien at personverninteressene må veies opp mot andre interesser, for eksempel interessen i helsehjelp, økonomiske interesser, samfunnets interesser i effektiv utnyttelse av felles ressurser mv. Personverninteressene representerer med andre ord kun slike hensyn som det kan/bør tas hensyn til når avveininger vedrørende personvern skal skje, for eksempel ved den enkelte helseforskers utforming av forskningsprosjekter som innebærer behandling av personopplysninger, og ved avgjørelse av konsesjonssøknad etter personopplysningsloven.
Nedenfor er personverninteressene regnet opp. I tilknytning til de to første interessene er det dessuten gjengitt krav som kan sies å øke sannsynligheten for at interessene skal kunne ivaretas. I faglitteraturen er det også spesifisert krav under de øvrige tre, men disse kommer vi ikke nærmere inn på i denne generelle gjennomgangen av personverninteressene. 75 Alle interesser har fått til dels stort gjennomslag i dagens lovgivning, spesielt i lovgivning som særskilt regulerer behandling av personopplysninger. I den følgende gjennomgangen vil det bare bli nevnt enkelte bestemmelser som kan illustrere hvordan interessene kan ivaretas.
Interessen i selvbestemmelse
Krav om råderett over egne personopplysninger
Krav om etablert tillitsforhold
Krav om konfidensialitet
Krav om beskyttet privatliv
Krav om vern av individets identitetsbilde
Interessen i innsyn og kunnskap
Krav om rettsinformasjon
Krav om generelt innsyn i systemer og rutiner for behandling av personopplysninger
Krav om individuelt innsyn i opplysninger om seg selv
Krav om begrunnelse for at personopplysninger er holdbare
Interessen i opplysnings- og behandlingskvalitet
Interessen i forholdsmessig kontroll
Interessen i brukervennlig behandling
Interessen i selvbestemmelse kan på mange måter ses som en overordnet interesse. Samtidig er det på det rene at denne interessen i mange tilfelle ikke kan realiseres fullt ut. Interessen i selvbestemmelse innebærer at den enkelte bør ha råderett over egne personopplysninger, noe som bl.a. kommer til uttrykk i krav til samtykke fra den registrerte flere steder i lovgivningen. Når slikt samtykke er gitt, eller når opplysningene behandles uten samtykke, innebærer denne interessen at behandlingen i så stor grad som mulig skal være basert på et tillitsforhold mellom den registrerte og den som behandler opplysningene. Tillitsskapende tiltak er således viktig for ivaretakelsen av interessen i selvbestemmelse dersom det ikke er mulig å gi full råderett. Bestemmelsesrett knyttet til tilgang til personopplysninger har en fremtredende plass i forhold til selvbestemmelsen. Den enkelte bør ha så stor grad av bestemmelsesrett som mulig over hvem som skal få tilgang til opplysninger om ham eller henne, jf kravet om konfidensialitet. Kravet om beskyttelse av privatliv gjelder særlig beskyttelse av private områder og personlige relasjoner innen hushold, familier (slekt) og vennekretser. Den enkelte må selv kunne bestemme hvor åpen vedkommende ønsker å være. Gjelder det relasjonene innen et hushold eller lignende, bør det være enighet blant de som inngår i denne kretsen. Kravet til vern om individets identitetsbilde gjelder personers evne til selv å bestemme over hvordan de fremstår både i forhold til andre og til seg selv. Personer kan være uvitende om eller ha fortrengt visse opplysninger om seg selv. Dersom andre har samlet inn eller generert opplysninger om slike forhold, kan åpenbaring av opplysningene forstyrre selvbildet og volde psykisk skade. En slik situasjon vil bl.a. kunne oppstå i forhold til åpenbaring av psykologiske eller medisinske testresultater. Bioteknologiloven § 6a-1 fjerde og femte ledd som begrenser adgangen til å drive oppsøkende genetisk veiledning, kan ses om utslag av dette kravet, jf. også Oviedo-konvensjonen 76 artikkel 10 andre ledd som nedfeller en rett til ikke å vite om opplysninger om egen helse:
Everyone is entitled to know any information collected about his or her health. However, the wishes of individuals not to be so informed shall be observed.
Interessen i innsyn og kunnskap er en viktig forutsetning for at den enkelte skal kunne ivareta sine interesser i selvbestemmelse. Kravet om rettsinformasjon gjelder både krav om at den enkelte skal kunne få kjennskap til hvilke rettigheter de har, og hvilke plikter den som behandler personopplysninger om dem har. Kravet om generelt innsyn gjelder retten til å få kjennskap til hvilke rutiner og systemer som blir benyttet ved behandling av personinformasjon, og de nærmere forholdene knyttet til informasjonsbehandlingen som kan ha betydning for ivaretagelse av den enkeltes personvern. Hvilke opplysningstyper og kilder som blir benyttet, samt hvem opplysninger eventuelt blir utlevert til, er sentrale eksempler på relevante forhold. Nært knyttet til krav om generelt innsyn er kravet om individuelt innsyn, dvs kravet om at den enkelte skal kunne få kjenneskap til opplysninger om ham eller henne; så vel faktiske opplysninger som vurderinger. Kravet om begrunnelse gjelder forklaringer på hvorfor en personopplysning har et bestemt innhold. Hvis personopplysningen går ut på at en person har disposisjon for en bestemt sykdom, innebærer dette kravet for eksempel at det bør gis forklaring på hvorfor denne opplysningen er riktig, noe som gjør at det må redegjøres for det resonnementet som leder frem til konklusjonen.
Interessen i opplysnings- og behandlingskvalitet innebærer kvalitetskrav på i alle fall tre nivåer. Personopplysninger skal være en dekkende representasjon av den person som blir beskrevet, opplysningene skal ha en kvalitet som står i forhold til bruksformålet, og den samlede behandlingen av alle opplysninger skal skje ved hjelp av informasjonssystemer og –rutiner som ivaretar kvalitetskravene. Når denne interessen settes inn i en forskningssammenheng, vil den langt på vei være dekket av aktuelle krav til forskningsmetode vedrørende innsamling og videre behandling av empiri knyttet til enkeltindivider.
Interessen i forholdsmessig kontroll stiller krav til bruk av kontrollmidler som motvirker ensidighet og uakseptabel utnyttelse av formelle/uformelle maktposisjoner overfor enkeltmennesker. Dersom det er knyttet helsehjelp til et forskningsprosjekt og denne helsehjelpen ses på som et knapphetsgode det kun er mulig å få tilgang til ved å akseptere rollen som forsøksperson, kan denne interessen aktualiseres.
Interessen i brukervennlig behandling gjelder særlig tilrettelegging for åpen og forståelig dialog mellom den som behandler personopplysninger og den enkelte som det er registrert opplysninger om, for eksempel mellom helseforsker og forsøkspersoner. Også spørsmål om driftsstabilitetet og sikring av lovlig tilgang til opplysninger, kan sies å høre inn under denne interessen. Uforståelige journalopplysninger og journaler som ikke er tilgjengelige pga systemfeil e.l. er eksempler på situasjoner som kommer i konflikt med denne interessen.
14.3 Helseregisterloven
14.3.1 Saklig virkeområde mv
Lov om helseregistre og behandling av helseopplysninger av 18. mai 2001 nr. 24 (helseregisterloven), gjelder for behandling av helseopplysninger innen helsesektoren, herunder innen forskning når denne gjør bruk av helseopplysninger. ”Helseopplysninger” er helseregisterloven § 2 nr. 1 definert som:
taushetsbelagte opplysninger i henhold til helsepersonelloven § 21 og andre opplysninger og vurderinger om helseforhold eller av betydning for helseforhold, som kan knyttes til en enkeltperson.
Helseregisterloven regulerer behandling av helseopplysninger i to hovedsituasjoner:
Når opplysningene behandles ved hjelp av elektroniske hjelpemidler, og
når helseopplysninger befinner seg i et helseregister eller skal inn i et slikt register.
Første situasjon gjelder elektronisk utstyr av enhver art, og omfatter således opplysninger i form av skrift, bilde, lyd og ethvert annet uttrykk for saksforhold eller vurderinger som er knyttet til en fysisk person. Det spiller ingen rolle hva behandlingen mer spesifikt består i; alt det en kan tenke seg å gjøre med helseopplysninger (innsamling, lagring, manipulering, analyse mv), faller inn under behandlingsbegrepet.
Andre situasjonen gjelder manuell behandling av helseopplysninger, men her stilles det krav til at opplysningene må være / vil bli organisert i et ”helseregister”, dvs på en systematisk måte som letter gjenfinning av opplysninger om den enkelte. Et manuelt kartotek med røntgenbilder kommer for eksempel inn under loven, og i tillegg alle bilder som skal inngå i et slikt kartotek.
Loven legger primært plikter på ”databehandlingsansvarlig”, 77 som er den som bestemmer formålet med behandlingen av opplysninger og hvilke hjelpemidler som skal brukes. Dette vil ofte være den øverste ledelsen av en virksomhet, for eksempel av den forskningsinstitusjon som står ansvarlig for gjennomføring av forskningsprosjektet. For behandlingsrettede helseregistre fastsetter imidlertid loven selv at databehandlingsansvaret skal ligge til ”Regionale helseforetak og helseforetak, kommune og annen offentlig eller privat virksomhet som tar i bruk behandlingsrettede helseregistre”. Tilsvarende er databehandlingsansvaret for regionale helseregistre lagt til det regionale helseforetaket, mens databehandlingsansvaret for sentrale helseregistre skal fastsettes i forskrift, jf § 8. 78 Databehandlingsansvar etter §§ 6, 7 og 8 kan delegeres, og det er ikke satt opp noen nærmere grense for slik delegasjon.
Hvilket geografisk område loven gjelder for, bestemmes av hvor den databehandlingsansvarlige er etablert . Er vedkommende etablert i Norge, gjelder helseregisterloven. Er databehandlingsansvarlige ikke etablert i Norge, men i et EØS-land, gjelder vedkommende EØS-lands lovgivning selv om behandlingen skjer i Norge. Datatilsynet kan anvende det aktuelle landets lov og også be om assistanse fra datatilsynsmyndigheter hos det landet der databehandlingsansvarlige er etablert. I mangel av særlovgivning for helseopplysninger i andre land, vil landenes generelle lovgivning vedrørende behandling av personopplysninger gjelde. Lovgivningen i EØS-land skal uansett være i overensstemmelse med personverndirektivet, noe som innebærer at det ikke vil være vesentlig avvik mellom annen lovgivning i EØS-land og helseregisterloven.
Dersom databehandlingsansvarlige er etablert utenfor EØS-området, gjelder ikke norsk lovgivning, med mindre det gjøres bruk av utstyr plassert i Norge som gjør mer enn å overføre helseopplysninger. Dersom en databehandlingsansvarlig er etablert i USA, men behandler opplysninger i Norge på substansielle måter, gjelder altså norsk lov. I et slikt tilfelle vil det i tillegg gjelde strenge regler for overføring av personopplysninger til ”tredjeland”, noe som i praksis kan gjøre det problematisk å overføre resultater av analyser mv fra Norge til USA og andre land som ikke anses å ha et tilfredsstillende sikkerhetsnivå for personopplysningene, se personopplysningsloven §§ 29 – 30.
14.3.2 Helseopplysningsbegrepet
Begrepet ”helseopplysning” er som nevnt en underkategori av ”personopplysning” som benyttes i personopplysningsloven (jf personverndirektivet), og de to begrepene skal grunnleggende avgrenses på samme måte. Forutsetningen for at en opplysning kan klassifiseres som en helseopplysning er at den – direkte eller indirekte – kan knyttes til en fysisk enkeltperson. Etter personopplysningsloven er det kun tale om opplysninger om levende personer. ”Helseopplysninger” favner videre. I den grad opplysningene er pasientopplysninger og underlagt helselovgivningens bestemmelser om taushetsplikt (jf helsepersonelloven § 21), vil helseopplysninger også omfatte opplysninger om døde personer. Dødsårsaksregisteret er for eksempel et helseregister over avdøde personer. Denne forskjellen innebærer bl.a at helseopplysninger om døde personer kan behandles uten direkte hensyn til personverndirektivet.
Det er muligheten for identifisering som er det avgjørende for spørsmålet om det foreligger en helseopplysning eller ikke. Således er alle opplysninger som kan knyttes til en bestemt person ”helseopplysninger” i lovens forstand, inkludert visse avidentifiserte 79 og alle pseudonyme opplysninger 80 . Dette betyr ikke at begrepet helseopplysning er ubegrenset. For det første faller anonyme opplysninger utenfor begrepet, dvs opplysninger der navn, fødselsnummer og andre personentydige kjennetegn ikke lenger kan knyttes til en enkeltperson, se helseregisterloven § 2 nr 3. Vi kommer tilbake til spørsmål om avidentifiserte, krypterte, pseudonymiserte og anonyme opplysninger straks nedenfor.
Noen opplysninger befinner seg i en gråsone for hva som kan anses å være ”helseopplysning”, dvs det kan være knyttet slike problemer til identifiseringen at opplysningene tenderer mot å måtte betraktes som anonyme. De to følgende avgrensningskriteriene er viktige når en i slike tvilstilfelle skal ta stilling til om det foreligger en ”helseopplysning” eller ikke.
For det første er det nødvendig å avgrense ut i fra sannsynligheten for at identifisering skal skje . Dersom identifisering er usannsynlig, er opplysningen ikke en ”helseopplysning” og helseregisterloven kommer ikke til anvendelse. Sannsynligheten skal bedømmes ut i fra ”alle hjelpemidler … som det er rimelig å ta i bruk for å identifisere vedkommende, enten av den databehandlingsansvarlige eller av en annen person” (se personverndirektivets fortale nr 26). Identifiseringsteknikkene kan med andre ord både være kostbare, tidkrevende og avanserte; opplysningene må uansett anses som ”helseopplysninger” så lenge det er trolig at de vil komme til å bli anvendt. Formålet med forskningsprosjektet samt generelle, eventuelt langsiktige forskningsmål / -forhåpninger / -behov er relevant når en skal ta stilling til dette spørsmålet i tilknytning til helserelatert forskning.
For det andre er det nødvendig å avgrense ut i fra hvor sikker identifisering som kan skje . Dersom identifiseringen er usikker, for eksempel dersom opplysningene alternativt kan gjelde flere enn 4 – 5 individer, kan det lett skje at opplysningene ikke ses som ”helseopplysninger”. Spørsmålet må imidlertid vurderes konkret, bl.a. ut i fra de aktuelle personvernmessige spørsmålene. Kravet om beskyttet privatliv kan tilsi at opplysninger bør anses som helseopplysninger når usikkerheten gjelder personer i samme familie, selv om antallet mulige personer er så høyt at vi uten familierelasjonen ikke vil anse opplysningen å være ”helseopplysning” i lovens forstand. Dersom en opplysning med omtrent samme sannsynlighet kan knyttes til ett av fem familiemedlemmer, kan opplysningen likevel anses å være ”helseopplysning”; derimot ikke hvis de fem mulige kandidatene utgjør en mer tilfeldig sammensatt gruppe.
Et avgjørende skille i helseregisterloven, personopplysningsloven og annen lovgivning som gjelder vern av personopplysninger, er altså om en opplysning lar seg knytte til en bestemt fysisk person eller ikke. Dersom opplysningene er identifiserbare, foreligger det ”helseopplysninger”, mens opplysninger som ikke på tilstrekkelige måter kan knyttes til en enkeltperson fordi det er usannsynlig at identifisering vil skje eller på grunn av for usikker identifisering (jf ovenfor), ikke regnes som identifiserbar. Dersom en helseopplysning ikke er identifiserbar, er den i prinsippet ”anonym”. Dette innebærer at vi må operere med ulike grader av anonymitet, på lignende måte som vi må operere med ulike grader av identifiserbarhet. I hvert ytterpunkt har vi tilfelle med utvilsom identitet og fullstendig uvisshet om identitet, mens vi omkring skillelinjen mellom en identifiserbar og en anonym opplysning må foreta en konkret vurdering. Også formålsbestemmelsen i helseregisterloven § 1 er viktig for slike konkrete vurderinger. Dette innebærer bl.a at en opplysning lettere kan anses å være anonym dersom det ikke er alvorlige personvernkonsekvenser knyttet til opplysningen. Dersom mulige personvernkonsekvenser er alvorlige, taler dette for å anse en opplysning som en ”helseopplysning”.
Skillet mellom identifiserbare og anonyme opplysninger, dvs mellom opplysninger som anses å være ”helseopplysninger” og slike opplysninger som faller utenfor dette begrepet, har meget stor rettslig betydning: Dersom det foreligger ”helseopplysninger” gjelder helseregisterloven, er opplysningene anonyme kan opplysningene behandles uten at denne loven kommer til anvendelse. I tillegg til hovedskillet mellom identifiserbar og anonym, finnes det flere teknikker for behandling av opplysninger som har betydning for dette skillet.
For det første kan opplysninger være kryptert . Det er særlig to hovedtilfelle av kryptering som er interessant for spørsmålet om det foreligger en ”helseopplysning” eller ikke, nemlig a) når selve opplysningen som kan knyttes til en person er kryptert, og b) når selve det identifiserende elementet (f.eks. personnummer) er kryptert. I tilfelle a) er det anvendt teknikker som gjør det semantiske innholdet av opplysningene vanskelig tilgjengelig for uvedkommende. 81 Situasjonen er for eksempel at en person med personnummer 12345678901 har diagnosen kreft, men denne diagnosen er kryptert og er derfor utilgjengelig. 82 Spørsmålet om anvendelse av kryptering har normalt ikke betydning for spørsmålet om det foreligger en ”helseopplysning” eller ikke. Opplysningen trenger mao ikke være lesbar for enhver for at opplysningen skal anses å være en ”helseopplysning”; det er tilstrekkelig at noen har tilgang til krypteringsnøkkelen og dermed kan dekryptere det semantiske innholdet av diagnoseopplysninger e.l..
Det neste tilfellet (b) gjelder situasjoner der det identifiserende elementet (f.eks. personnummeret) er kryptert eller skjult. Helseregisterloven betegner slike opplysninger ”pseudonyme helseopplysninger”, dvs ”helseopplysninger der identitet er kryptert eller skjult på annet vis, men likevel individualisert slik at det lar seg gjøre å følge hver person gjennom helsesystemet uten at identiteten røpes.” Utvalget mener at begrepet ”pseudonyme helseopplysninger” har vist seg å være vanskelig å forstå for brukerne, og anbefaler derfor at man i stedet taler om ”helseopplysninger med skjult identitet”, uten at meningsinnholdet endres, se kapittel 24. Mens vi ovenfor har omtalt tilfelle der det semantiske innholdet av en opplysning er kryptert, gjelder pseudonymiseringen tilfelle der det identifiserende elementet (f.eks. personnummeret) er kryptert eller erstattet med ”falsk” entydig identitet. Også i tilfelle av pseudonyme opplysninger, foreligger det ”helseopplysninger”, fordi det både er mulig og sikkert å finne frem til rett identitet for alle som får tilgang til ”nøkkelen”.
Helseregisterloven definerer dessuten noen helseopplysninger som ”avidentifiserte”, dvs ”helseopplysninger der navn, fødselsnummer og andre personentydige kjennetegn er fjernet, slik at opplysningene ikke lenger kan knyttes til en enkeltperson, og hvor identitet bare kan tilbakeføres ved sammenstilling med de samme opplysninger som tidligere ble fjernet”. Definisjonen av avidentifisering i helseregisterloven forutsetter at identitetene er fjernet på en måte som legger til rette for at identitetene kan tilbakeføres uten at det oppstår feil. Selv om denne forutsetningen holder stikk, er avidentifisering mindre sikker enn pseudonymisering, fordi pseudonymisering forutsetter kryptering av det identifiserende elementet Ved kryptering og dekryptering vil det bli utført regneoperasjoner som innebærer sikrere overganger tilbake til virkelige identiteter enn hva tilfellet er for reetablering av identiteter der opplysningene er avidentifiserte. Dersom en ikke i tilstrekkelig grad har forberedt rutiner for reetablering av avidentifiserte opplysninger, vil det kunne oppstå feil ved tilbakeføringen. For at avidentifisering skal kunne anbefales, må det derfor stilles klare og strenge krav til fremgangsmåtene.
Den siste distinksjonen som skal nevnes her, er skillet mellom ”helseopplysning” og humant biologisk materiale. I biobankloven er ”humant biologisk materiale” definert som ”organer, deler av organer, celler og vev og bestanddeler av slikt materiale fra levende og døde mennesker”. Det er altså intet opplysningselement knyttet til materialet i seg selv, og materialet defineres ikke som ”helseopplysning”. I definisjonen av ”forskningsbiobank” i samme lov, er slike banker definert som ” samling humant biologisk materiale og opplysninger som direkte fremkommer ved analyse av dette materialet [...]” (vår kursiv). Det er uklart hva dette innebærer. Det selvsagte er at en kan trekke slutninger om mennesker på grunnlag av en analyse av slikt materiale, og det skjelnes ikke mellom ulike analysemetoder. 83 Foruten materialet selv er bare slike opplysninger som direkte fremkommer ved analyse, del av biobanken. Blant slike opplysninger kan være opplysninger om identitet, men trenger ikke være det. Som oftest vil imidlertid selve det organiske materialet være identifisert ved uttaket .
”Forskningsbiobank” betegner en kombinasjon av humant biologisk materiale og avledede opplysninger. Dersom materialet er knyttet til en bestemt person ved uttak eller gjennom senere analyse av materiale, vil biobanken inneholde ”helseopplysninger”. Humant biologisk materialet er ikke en ”helseopplysning” i seg selv, men prinsipielt en bærer av slike opplysninger. 84 Et hårstrå i frisørsalongen er derfor ingen ”helseopplysning”, men de opplysninger som kan knyttes til hårstrået og som fremkommer i forbindelse med uttak og/eller analyse av materialet, må anses å være ”helseopplysning” i lovens forstand. 85
14.3.3 Rettslig grunnlag for å behandle helseopplysninger
For at det overhode skal være anledning til å behandle helseopplysninger, må det foreligge et rettslig grunnlag for behandlingen, jf. helseregisterloven § 5, som må forstås slik at det bare er anledning til å behandle helseopplysninger etter helseregisterloven på grunnlag samtykke fra den personen opplysningene gjelder eller på grunnlag av lovhjemmel.
Helseregisterloven §§ 7 og 8 begrenser adgangen til å opprette nye lokale, regionale og sentrale helseregistre. For at slike registre skal kunne opprettes, må det både finnes en forskriftshjemmel, og hver enkelt person som det skal behandles opplysninger om, må samtykke til at databehandlingen kan finne sted. Samtykke er likevel ikke nødvendig dersom det i forskrift er bestemt at registrene bare skal inneholde pseudonymiserte og/eller avidentifiserte opplysninger.
Lovgiver kan selvsagt vedta opprettelse av nye helseregistre. Uten slikt lovvedtak kan opprettelse av nye lokale, regionale og sentrale helseregistre bare skje ved forskrift vedtatt av Kongen i statsråd i samsvar med helseregisterloven §§ 7 og 8. Kongens forskriftskompetanse er begrenset til helseregistre som ivaretar oppgaver etter bestemte helselover. De aktuelle lovene er regnet opp i helseregisterloven §§ 7 og 8 og varierer avhengig av om helseregisteret er lokalt, regionalt eller sentralt.
Helseregisterlovens krav til samtykke er fastlagt i helseregisterloven § 2 nr 11 der det stilles krav til selve viljeserklæringen. Bestemmelsen er i samsvar med en tilsvarende bestemmelse i personopplysningsloven § 2 nr 7. I begge bestemmelser kreves det at samtykket skal være informert, frivillig og uttrykkelig. At samtykket er informert , innebærer trolig at den enkelte skal kjenne til de saksforhold som den databehandlingsansvarlige alltid skal informere om etter helseregisterloven §§ 23 og 24. I tillegg bør det informeres om åpenbare og /eller sannsynlige effekter av behandlingen for den aktuelle personens personvern; for eksempel at selvbildet vil kunne bli rokket på grunn av forskningsresultatene, jf helseregisterloven § 22 tredje ledd. Kravet om frivillighet innebærer at den enkelte ikke er utsatt for et slikt press at det ikke eksisterer en reell valgsituasjon. Kravet om at samtykket skal være uttrykkelig , innebærer intet formkrav, men samtykket skal komme til syne gjennom en aktiv handling som kan ettervise at samtykket er avgitt. I forskningssammenheng bør det normalt innebære et krav om skriftelighet.
Kravene til samtykke etter helseregisterloven suppleres av samtykkebestemmelser i pasientrettighetsloven §§ 4-3 – 4-8. Disse bestemmelsene regulerer spørsmålet om hvem som har samtykkekompetanse på vegne av mindreårige, andre umyndige personer og myndige personer som ikke kan avgi samtykke pga senilitet, mentale forstyrrelser mv. Tilsvarende bestemmelser finnes ikke i eller i medhold av personopplysningsloven.
Helseopplysninger kan som nevnt behandles dersom det er hjemlet i eller i medhold av lov. Hva det faktisk er hjemmel for å foreta av behandling, beror på en konkret fortolkning. Utgangspunktet er imidlertid at hjemmelen må være eksplisitt. Implisitt hjemmel kan tenkes å foreligge dersom det i lov er beskrevet oppgaver som bare kan utføres ved å behandle bestemte personopplysninger. Dersom det er knyttet vesentlige personvernbetenkeligheter til slik databehandling, er det imidlertid grunn til å stille strenge krav til hjemmelsgrunnlaget.
Som tidligere nevnt er det forskjeller mellom krav til rettslig grunnlag for å behandle helseopplysninger etter personopplysningsloven og helseregisterloven. Etter helseregisterloven er det kun samtykke og lovhjemmel som er mulige grunnlag. Etter personopplysningsloven er det ytterligere to mulige hjemmelsgrunnlag:
Dersom en databehandlingsansvarlig ikke kan vise til lovhjemmel eller samtykke som rettslig grunnlag for behandlingen av personopplysninger, er neste mulighet i personopplysningsloven at det foreligger ”nødvendighet” slik det er fastsatt i personopplysningsloven § 9 første ledd, bokstavene c og e - h. I samband med medisinsk forskning er de følgende alternativene særlig aktuelle:
”c) beskytte en persons vitale interesse”,
”g) forebyggende sykdomsbehandling, medisinsk diagnose, sykepleie eller pasientbehandling [mv]” og
”h) [bl.a.] vitenskapelige formål” som etter en helhetsvurdering representerer samfunnsinteresser som ”klart overstiger ulempene den kan medføre for den enkelte”.
Det siste mulige rettslige grunnlaget er at ”det utelukkende behandles opplysninger som den registrerte selv frivillig har gjort alminnelig kjent”, se personopplysningsloven § 9 bokstav d. Dersom det bare foreligger et slikt rettslig grunnlag, er det altså ikke lov å kombinere behandling av disse personopplysningene med andre personopplysninger.
I utgangspunktet er det den databehandlingsansvarlige selv som må ta stilling til om det eksisterer et holdbart rettslig grunnlag. I de fleste tilfelle der helseopplysninger skal behandles, vil det foreligge en plikt til å innhente konsesjon (forhåndsgodkjennelse) fra Datatilsynet på den planlagte behandlingen av personopplysninger, se avsnitt 14.3.8. Det rettslige grunnlaget vil selvsagt også bli prøvet av Datatilsynet som ledd i konsesjonsbehandlingen.
14.3.4 Angivelse av formål for databehandling av helseopplysninger
Det er ikke tillatt å behandle helseopplysninger med mindre det på forhånd er angitt ett eller flere formål som er saklig begrunnet i virksomheten, se helseregisterloven § 11 første ledd. Det er imidlertid ikke angitt krav til hvor detaljert formålsbeskrivelsen må være, og generelt er det grunn til å anta at faglig akseptable beskrivelser av forskningsprosjekter vil tilfredsstille kravet til formålsbeskrivelse. Formålet kan endres, men ikke på en slik måte at det nye formålet er uforenelig med det opprinnelige formålet. Det er derfor mulig at et forskningsprosjekt som i utgangspunktet skulle forske på antatt uforsvarlig medisinsk behandling av en pasientgruppe, ikke kan endre eller supplere problemstilling underveis og i stedet undersøke hvorvidt denne pasientgruppen kan sies å ha forårsaket sin egen sykdomssituasjon. Trolig skal det imidlertid mye til for at et nytt formål kommer i slik konflikt med det opprinnelige formålet, at endringen må nektes.
Dersom formålet med å behandle helseopplysninger er annet enn helsehjelp og/eller administrasjon av helsehjelp, må det alltid begrunnes hvorfor det er nødvendig å gjøre bruk av personidentifiserbare opplysninger, se helseregisterloven § 11 annet ledd, og begrunnelsen kan kreves fremlagt for tilsynsmyndighetene. ”Helsehjelp” må formodentlig forstås på samme måte som i pasientrettighetsloven § 1-3 bokstav c, noe som innebærer at begrepet normalt ikke omfatter forskning. I tilknytning til helseforskning må man derfor alltid begrunne hvorfor det er nødvendig å benytte personidentifiserbare opplysninger.
14.3.5 Informasjonssikkerhet, opplysningskvalitet og internkontroll
Helseregisterloven § 16 stiller krav til sikring av helseopplysningers konfidensialitet, integritet og tilgjengelighet. Sikkerhetsnivået skal være ”tilfredsstillende”, noe som innebærer en konkret vurdering av risiko. Loven selv stiller ingen nærmere krav til sikkerhetsarbeidet, men detaljregler gis i kapittel 2 i personopplysningsforskriften, som i medhold av helseregisterloven § 36 gjelder som utfyllende bestemmelse. Her stilles det opp en rekke generelle krav til sikkerhetsarbeidet, som i konkrete tilfelle kan sies å være mer vidtrekkende enn det som kreves for å sikre ”tilfredsstillende” sikkerhet. Forskriftsbestemmelsen må trolig leses med denne reservasjonen, for å hindre at forskriften gis et innhold som går ut over hjemmelen i helseregisterloven § 16.
Som en del av bestemmelsen om informasjonssikkerhet, er det også tatt med et krav til at helseopplysninger skal ha tilfredsstillende kvalitet. Heller ikke her gir loven noen nærmere holdepunkter for det nærmere innholdet av kravet, og heller ikke personopplysningsforskriften inneholder slike nærmere anvisninger. Fordi personopplysningsloven er utfyllende (jf helseregisterloven § 36), må kravet leses i samband med personopplysningsloven § 11 bokstavene d og e som stiller krav til at opplysningene skal være tilstrekkelige, korrekte, oppdaterte og ikke bli lagret lengre enn formålet tilsier. Kvalitetskravet må også forstås i lys av helseregisterloven § 17 første ledd, som spesielt stiller krav til arbeidet med opplysningskvalitet i tilknytning til arbeidet med internkontroll.
Internkontrollarbeidet skal sikre at det blir iverksatt slike tiltak som er nødvendige for å oppfylle krav som er fastsatt i eller i medhold av loven. Dette gjelder for det første krav som følger av helseregisterloven med forskrifter, samt utfyllende regler i personopplysningsloven med forskrifter. Dessuten gjelder kravet til internkontroll for alle konsesjonsvedtak, og andre enkeltvedtak som gjelder vedkommende behandling av personopplysninger. På den annen side gjelder ikke kravet til internkontroll krav som er hjemlet i annet enn helseregisterloven mv. Således skaper ikke helseregisterloven § 17 noen plikt til å la internkontrollsystemet omfatte de regionale komiteer for medisinsk forskningsetikk. kapittel 3 i personopplysningsforskriften om internkontroll gjelder for behandling av helseopplysninger som utfyllende bestemmelser. Kapittelet er langt mindre omfattende enn kapittel 2 om informasjonssikkerhet, men også her gis det pålegg om tiltak som kan komme i konflikt med lovens forutsetning om konkret vurdering av behovet for internkontroll (”nødvendig”), jf forskriftens § 3-1 tredje ledd bokstavene a – f.
Alle tiltak som er knyttet til arbeidet med informasjonssikkerhet, opplysningskvalitet og internkontroll (helseregisterloven §§ 16 og 17), skal være planlagte, systematiske og dokumenterte. Kravet til planlegging innebærer at de må være gjennomført på forhånd og i samsvar med en helhetlig plan. Kravet til systematiske tiltak innebærer at tiltakene må være resultat av en systematisk forståelse, gjerne ved at det blir anvendt en konkret metodikk. Dokumentasjonskravet innebærer at tiltakene skal være beskrevet på en måte som gjør det mulig for tilsatte og tilsynsmyndigheter å sette seg inn i hva tiltakene faktisk går ut på. Dersom behandling av personopplysninger settes bort til en ”databehandler” (”outsourcing”), det vil si en som behandler personopplysninger på vegne av den databehandlingsansvarlige, skal det alltid inngås en skriftlig avtale som beskriver databehandlers disposisjon over helseopplysningene, og som forplikter oppdragstaker til å gjennomføre de sikringstiltakene som er beskrevet i dokumentasjonen. Også dokumentasjonen vedrørende internkontroll skal være tilgjengelig for medarbeidere hos databehandler.
14.3.6 Sammenstilling og utlevering av helseopplysninger
I personopplysningsloven er spørsmålet om sammenstilling (”kopling”/”samkjøring”) av personopplysninger ikke spesielt regulert, men adgangen til å sammenstille opplysninger vil avhenge av om det foreligger et tilstrekkelig rettslig grunnlag, angivelsen av formålet med behandlingen mv. Helseregisterloven angir eksplisitte regler om slik sammenstilling i § 12. 86 I første ledd bestemmes det at ”behandlingsrettede helseregister” bare kan sammenstilles med opplysninger om samme pasient når helseopplysningene kan utleveres etter helsepersonelloven §§ 25, 26 og 45. Behandlingsrettede helseregistre er betegnelse på pasientjournaler og andre helseregistre og informasjons-systemer som benyttes av helsepersonell i den medisinske behandlingen av den enkelte pasient (forebyggende, diagnostisk, behandlende, helsebevarende og rehabiliterende), samt administrasjonen av slik medisinsk behandling.
Helsepersonelloven §§ 25 og 26 gjør unntak fra taushetsplikten etter helsepersonelloven § 21, og tillater utlevering av opplysninger til samarbeidende helsepersonell og til pasientadministrasjon, internkontroll og kvalitetssikring av helsetjenester. Ingen av disse bestemmelsene hjemler utlevering for forskningsformål. Etter helsepersonelloven § 29 kan departementet bestemme at helseopplysninger skal eller kan benyttes til forskning uten hinder av taushetsplikt. Helseregisterloven viser imidlertid ikke til helsepersonelloven § 29. Det er trolig at dette skyldes en inkurie, og spørsmålet er ikke nevnt i de særlige merknadene til bestemmelsen, se Ot.prp. nr. 5 (1999 – 2000). For slike helseregistre er det i tillegg fastlagt at de kan sammenstilles med folkeregisteropplysninger om den registrerte.
Når det gjelder sammenstilling av helseopplysninger som er samlet inn til lokale, regionale og sentrale helseregistre (helseregisterloven §§ 7 og 8, jf § 9), kan dette bare skje i den grad det er tillatt i den forskriften som skal regulere vedkommende register, jf nedenfor. Av de åtte aktuelle forskriftene om sentrale helseregistre, har seks forskrifter eksplisitte bestemmelser om adgangen til å sammenstille helseopplysninger.
Tredje ledd av helseregisterloven § 12 bestemmer at sammenstilling av helseopplysninger i andre tilfelle enn de som er regulert i de to foregående leddene (jf ovenfor), bare kan skje ”når dette er tillatt etter personopplysningsloven §§ 9 og 33”. Denne ordlyden er misvisende fordi disse bestemmelsene ikke regulerer sammenstilling av opplysninger, men kun kan få betydning for spørsmålet om lovlighet. Imidlertid kan også andre bestemmelser i samme lov, og bestemmelser i helseregisterloven selv, ha betydning for lovligheten av slik sammenstilling. Poenget er at personopplysningsloven ikke behandler sammenstilling spesielt, og det er derfor lovens alminnelige bestemmelser som blir avgjørende for om sammenstilling av helseopplysninger er lovlig eller ikke. Henvisningen til personopplysningsloven § 9 er trolig ment som en påminnelse om at sammenstillingen må ha et rettslig grunnlag. Det er imidlertid også et vilkår at sammenstillingen kan skje innenfor det definerte formålet med behandlingen, og at opplysningene har den kvalitet som formålet gjør nødvendig. Det er videre et vilkår at opplysningene er sikret på tilfredsstillende måte, for eksempel slik at opplysningsintegriteten er ivaretatt for å unngå forveksling av identitet mv, jf personopplysningsloven § 12 (særlig annet ledd) om bruk av entydige identifikasjonsmidler.
Henvisningen i helseregisterloven § 12 til personopplysningsloven § 33 er trolig ment som en påminnelse om at helseopplysninger ofte er underlagt konsesjonsplikt. Ved en konsesjonsbehandling må Datatilsynet først ta stilling til lovligheten av sammenstillingen, bl.a i forhold til bestemmelser om taushetsplikt i helsepersonelloven kapittel 5, vilkårene for sammenstilling i helseregisterloven § 12 og de alminnelige kravene til behandling av helseopplysninger og personopplysninger ellers i helseregisterloven og personopplysningsloven. Forutsatt at sammenstillingen av opplysninger kan være lovlig, vil Datatilsynet foreta en avveining der personverninteressene vurderes opp mot de hensyn som begrunner sammenstillingen, se personopplysningsloven § 34. Tilsynet kan i denne sammenheng stille nærmere vilkår for at samkjøring kan skje, for eksempel stille krav om sikrere identifisering av den enkelte enn hva som er skissert i konsesjonssøknaden, jf personopplysningsloven § 35.
Enkelte behandlinger av helseopplysninger er unntatt fra konsesjonsplikten, blant disse er visse forskningsprosjekter, se personopplysningsforskriften § 7 – 27. Unntak gjelder imidlertid ikke dersom det skal foretas elektronisk sammenstilling av opplysninger, se § 7- 27 bokstav e.
Utlevering av helseopplysninger kan i henhold til helseregisterloven § 14 skje når:
opplysningene lovlig skal sammenstilles i henhold til helseregisterloven § 12, eller
opplysningene skal overføres for avidentifisering eller anonymisering, eller
utlevering av opplysningene har hjemmel i lov eller forskrift.
I alle tre situasjoner er det en forutsetningen at utleveringen ikke strider mot taushetsplikt og at den opplysningene utleveres til, har rett til å behandle opplysningene i henhold til bestemmelsene i helseregisterloven og personopplysningsloven. Det siste innebærer at den det utleveres helseopplysninger til, må ha et rettslig grunnlag, ha fastsatt formål for behandlingen, ha nødvendig konsesjon mv. Helseopplysninger som er resultat av en sammenstilling, kan utleveres når dette skjer for å avidentifisere eller anonymisere opplysningene. I slike tilfelle er det dessuten et vilkår at navn og fødselsnummer er fjernet før overføring skjer, og at utleveringen skjer til en virksomhet som er bestemt av departementet.
I følge forarbeidene er anvisning på rutiner for avidentifisering og anonymisering spesielt fastsatt for å legge til rette for videre overføring av opplysninger til forskningsformål. Avidentifiserte opplysninger er helseopplysninger i lovens forstand, og er derfor ikke fritatt fra konsesjon og lovens øvrige krav til databehandling av helseopplysninger, fordi det i slike tilfelle er mulig å reetablere identitetene, se definisjonen i helseregisterloven § 2 nr 2. Når opplysningene er anonymisert, er slik reetablering av identiteter ikke mulig, og den videre behandlingen av opplysningene er derfor ikke regulert av helseregisterloven eller personopplysningsloven, se helseregisterloven § 2 nr 3.
14.3.7 Retting og sletting av helseopplysninger
For behandlingsrettede helseregistre (jf definisjonen i helseregisterloven § 2 nr 7), gjelder bestemmelsene om retting og sletting av journal i helsepersonelloven kapittel 8. For øvrig gjelder bestemmelsene i helseregisterloven §§ 26 og 27 om retting, sletting mv og lagring av unødvendige helseopplysninger. I den grad det ikke oppstår motstrid mellom lovbestemmelsene, supplerer helseregisterlovens bestemmelser bestemmelsene i helsepersonelloven.
Helseregisterloven § 26 tar for seg tilfelle der helseopplysninger lider av en av tre typer mangler, nemlig når opplysningene er
uriktige,
ufullstendige eller
det ikke er adgang til å behandle opplysningene.
Manglende adgang til å behandle opplysningene kan for eksempel skyldes at det rettslige grunnlaget mangler, fordi det ikke er innhentet gyldig samtykke eller fordi samtykket er trukket tilbake. Det kan også være at det ikke er fastsatt noe formål for databehandlingen.
Loven tar direkte bare stilling til hva som skal skje med uriktige og ufullstendige helseopplysninger. Hovedregelen er at disse opplysningene skal rettes og suppleres, og at uriktige opplysninger skal slettes.
Dersom de mangelfulle opplysningene kan ha betydning for dokumentasjon, skal mangelfulle opplysninger suppleres og ukorrekte opplysninger rettes, samtidig som rettingen tydelig skal markeres. En uriktig opplysning skal markeres som uriktig, samtidig som korrekt opplysning føyes til, mens en ufullstendig opplysning føyes til, samtidig som det markeres at supplering har skjedd.
Dersom ”tungtveiende personvernhensyn tilsier det” kan Datatilsynet likevel kreve at uriktige og ufullstendige opplysninger sperres eller slettes. Dersom det er umulig å gi korrekte og fullstendige opplysninger, og de resterende opplysningene gir et gir et åpenbart misvisende bilde, skal hele dokumentet slettes. Loven inneholder ingen definisjon av ”dokument”, og det kan være tvil om hva som skal slettes dersom opplysningene for eksempel inngår i rapporter med vedlegg, videoopptak, databaser mv. Før sletting eller sperring skjer, skal Riksarkivaren uttale seg, men det er Datatilsynet som har kompetanse til å avgjøre saken.
Den databehandlingsansvarlige har plikt til å rette og slette mv opplysninger av eget tiltak, så snart hun eller han blir klar over manglene. Dersom den registrerte krever det, oppstår det også en plikt til å handle, men det kan neppe antas at det i slike tilfelle er en ubetinget plikt til å rette og slette dersom det er uenighet om hva som er korrekte og fullstendige opplysninger. Supplering som angir en uenighet, vil i slike tilfelle være en mulighet. Plikten til å rette mv etter anmodning fra den registrerte, er ikke i loven begrenset til tilfelle der den registrerte har innsynsrett.
Dersom det er oppdaget feil eller mangler ellers, plikter den databehandlingsansvarlige å treffe de tiltak som er nødvendige for å hindre at feilen får betydning for den registerte. Er opplysningene utleverte, skal den databehandlingsansvarlige varsle mottakere av opplysningene om feilen. Det fremgår imidlertid ikke av loven om aktivitets- og varslingsplikten inntreffer når det er på det rene at sikkert foreligger en feil/mangel.
Helseregisterloven § 27 regulerer tilfelle der helseopplysningene forutsettes å være fri for mangler, men der formålet med behandlingen av opplysningene ikke lenger tilsier behandling. Hovedregelen er i slike tilfelle at opplysningene enten skal overføres til Arkivverket eller slettes. 87 Når opplysningene er ferdig behandlet i forhold til det formålet de er innsamlet for, har den databehandlingsansvarlige og vedkommendes eventuelle databehandlere med andre ord ikke lenger adgang til å behandle helseopplysningene. I helseregisterloven § 27 annet ledd er det likevel gitt hjemmel for å fastsette vilkår for fortsatt lagring av helseopplysninger, dersom dette er ønskelig ut i fra (bl.a.) vitenskapelige formål. Forutsetningen er at samfunnets interesser i fortsatt lagring klart overstiger de ulemper slik lagring kan utgjøre for den enkelte. Siden slike bestemmelser må skje i forskrifts form (av Kongen i statsråd), vil det her være tale om generelle vurderinger som anses å ha gyldighet over forholdsvis lange tidsrom. Siden det sjelden er tilgjengelige forarbeider til forskrifter, må trolig interesseavveiningene – direkte eller indirekte – fremgå av selve forskriften, for på den måten å legge til rette for legalitetskontroll.
14.3.8 Innsyn og informasjon
Helseregisterloven kapittel 4 inneholder fire typer bestemmelser som er ment å skape åpenhet rundt behandling av helseopplysninger:
Plikt til å gi informasjon om databehandling av lokale, regionale og sentrale helseregistre til allmennheten, se § 20, jf §§ 7 og 8.
Rett til innsyn i visse generelle opplysninger knyttet til hver behandling av helseopplysninger (”generelt innsyn”), § 21.
Rett til innsyn for registrerte personer i opplysninger om dem selv (”individuelt innsyn”), § 22.
Plikt til å gi informasjon til personer det blir innhentet opplysninger fra, §§ 23 og 24.
Helseregisterloven § 20 innebærer at databehandlingsansvarlige for lokale, regionale og sentrale helseregistre som er etablert med hjemmel i helseregisterloven §§ 7 og 8, har plikt til aktivt å informere allmennheten om hva slags databehandling av helseopplysninger som foretas. En slik plikt gjelder ikke ved databehandling av andre helseopplysninger (jf §§ 5 og 6), og følger heller ikke av personopplysningsloven eller personverndirektivet. Bestemmelsen i § 20 fastsetter ikke noe nærmere om krav til innholdet av denne informasjonen, men i forarbeidene er det uttalt at informasjonen bør omfatte de samme opplysninger som alle har krav på i medhold av § 21 om ”generelt innsyn”. Dette innebærer for eksempel at sentrale helseregistre må informere om den behandling av personopplysninger de gjør innenfor rammen av forskningsprosjekter, og i tillegg gi flere andre opplysninger som her vil bli beskrevet straks nedenfor. Informasjonen er i forarbeidene gitt gjennom brosjyrer, aviser, TV, Internett etc.
Retten til generell informasjon, dvs til å kreve innsyn i ulike generelle forhold knyttet til informasjonsbehandlingen, gjelder for enhver, se helseregisterloven § 21. Retten er knyttet til en bestemt type behandling av helseopplysninger, for eksempel behandling som skjer innen et konkret forskningsprosjekt, og omfatter:
navn og adresse på den databehandlingsansvarlige og dennes eventuelle representant,
hvem som har det daglige ansvaret for å oppfylle den databehandlingsansvarliges plikter,
formålet med databehandlingen av helseopplysningene,
beskrivelser av hvilke typer helseopplysninger som behandles,
hvor opplysningene er hentet fra, og
om helseopplysningene vil bli utlevert, og eventuelt hvem som er mottaker.
Informasjonen kan både kreves fra den (data) behandlingsansvarlige og hos dennes eventuelle databehandler.
Retten til individuelt innsyn etter helseregisterloven § 22 er knyttet til innsynsbestemmelsene i pasientrettighetsloven § 5-1 (som angir innsynsretten) og helsepersonelloven § 41 (som angir helsepersonellets plikt til å medvirke til å realisere innsynsretten). Bestemmelsen i prl § 5-1 gir pasienten rett til innsyn i ”journalen sin med bilag”. Formuleringen er tilpasset situasjoner der helsepersonell gir helsehjelp og det således foreligger journalføringsplikt etter helsepersonelloven § 40, jf helseregisterloven § 6 og uttrykket ”behandlingsrettet helseregister”.
Helseregisterloven § 22 annet ledd gjelder innsyn i situasjoner der behandlingen skjer i et lokalt, regionalt eller sentralt helseregister (jf §§ 7 og 8), og i annen databehandling av helseopplysninger etter § 5, dvs utenfor behandlingsrettet helseregister. I slike tilfelle gjelder innsynsretten
hvilke helseopplysninger om den registrerte som behandles, og
sikkerhetstiltakene ved databehandlingen av helseopplysningene så langt innsyn ikke svekker sikkerheten.
Uansett om det er krevd innsyn etter første eller annet ledd, har den enkelte i tillegg krav på å få utdypet den informasjonen alle har krav på etter helseregisterloven § 21. Forutsetningen er at informasjonen er nødvendig for at den registrerte skal kunne ivareta egne interesser, men den databehandlingsansvarlige kan neppe i særlig grad overprøve den registrertes egen bedømmelse av behov for kunnskap. Pårørende, foreldre, eventuelt en representant for den registrerte, kan ha rett til innsyn i opplysninger om personer som selv ikke kan vareta egne interesser, jf pasientrettighetsloven § 5-1 tredje ledd og fjerde ledd som viser til pasientrettighetsloven §§ 3-3 og 3-4. 88
Etter §§ 23 og 24 skal det også gis informasjon når det samles inn helseopplysninger. Bestemmelsene skjelner mellom situasjoner der opplysninger blir direkte samlet inn fra den registrerte selv (for eksempel en forsøksperson), og situasjoner der opplysningene blir innhentet fra andre enn vedkommende person, for eksempel et annet forskningsregister. Hvilken informasjon som skal gis er likt i begge situasjoner. Imidlertid gjelder det egne unntaksbestemmelser i sist nevnte situasjon.
Den informasjonen som skal gis, er i stor grad overlappende med den informasjonen alle har krav på i medhold av § 21:
navn og adresse på den databehandlingsansvarlige og dennes eventuelle representant,
formålet med behandlingen av helseopplysningene,
opplysningene vil bli utlevert, og eventuelt hvem som er mottaker,
det er frivillig å gi fra seg helseopplysningene, og
annet som gjør den registrerte i stand til å bruke sine rettigheter etter loven her på best mulig måte, som for eksempel informasjon om retten til å kreve innsyn, jf § 22, og retten til å kreve retting og sletting, jf §§ 26 og 28.
I punktene 1) – 5) ovenfor er det kun opplysning om frivillighet i nr 4) som ikke også inngår i bestemmelsen om individuelt innsyn i helseregisterloven § 22. Spørsmålet om frivillig avgivelse av helseopplysninger kommer på spissen i situasjoner der opplysningene blir samlet inn fra andre enn den registrerte selv. Informasjonen kan nemlig utsettes til utlevering skjer, jf helseregisterloven § 24 første ledd. I så fall vil det også være for sent for en registrert person som ønsker å motsette seg utlevering fordi han blir klar over at utleveringen er frivillig.
Det skal ikke gis informasjon etter §§ 23 og 24 dersom det er på det rene at den registrerte allerede kjenner den aktuelle informasjonen. Når kilden for informasjonen er en annen enn den registrerte selv, gjelder det dessuten unntak dersom det er umulig eller uforholdsmessig vanskelig å varsle, eller dersom innhenting av opplysningene uttrykkelig er fastsatt i lov. Således kan hjemmelen til utlevering i helseregisterloven § 12 trolig være tilstrekkelig hjemmel til at varsel etter helseregisterloven § 24 ikke er påkrevet. I forarbeidene antar departementet at bestemmelser om utlevering i forskriftene til helseregisterloven §§ 7 og 8 også utgjør en tilstrekkelig hjemmel for å oppnå unntak.
Med ett avvik gjelder det felles unntaksregler for alle de nevnte bestemmelsene om innsyn og informasjon i helseregisterloven § 25. Avviket gjelder unntak fra innsyn i behandlingsrettede helseregistre, der unntak må ha hjemmel i pasientrettighetsloven § 5-1. Bestemmelsene i helseregisterloven § 25 som regulerer unntak fra innsyn i alle andre behandlinger av helseopplysninger etter loven, tilsvarer meget langt på vei tilsvarende unntaksbestemmelse i personopplysningsloven § 23. De fleste av alternativene er lite aktuelle for situasjoner vedrørende helseforskning. Bestemmelsens alternativ nr 3 er imidlertid særlig relevant og omfatter opplysninger som: ”det må anses utilrådelig at den registrerte får kjennskap til, av hensyn til vedkommendes helse eller forholdet til personer som står vedkommende nær”. Bestemmelsen kan for eksempel være aktuell i tilfelle der helseopplysninger røper slektsforhold og helseforhold mv som den registrerte sannsynligvis ikke vil være tjent med å få kjennskap til.
Også i medhold av helseregisterloven § 25 nr 5 kan det unntas helseopplysninger fra innsyn og informasjon på en måte som er relevant for helseforskning. Dette gjelder dersom opplysningene er knyttet til interne, saksforberedende dokumenter som finnes hos den databehandlingsansvarlige og / eller dennes databehandler, og som ikke er utlevert til andre. I bestemmelsen er unntaket spesielt knyttet til ”tekst”, men dette kan neppe tas bokstavelig; trolig vil enhver type helseopplysninger kunne unntas i medhold av denne bestemmelsen, dvs uavhengig av om opplysningen kommer til uttrykk som tekst, bilde, lyd eller lignende. Dette unntaket betyr formodentlig at helseopplysninger som inngår i empirisk materiale og i foreløpige forskningsresultater kan unntas fra innsyn og informasjon, så lenge opplysningene ikke er kommunisert til andre utenfor den databehandlingsansvarliges og / eller databehandlers organisasjon.
14.3.9 Konsesjons- og meldeplikt
Helseregisterloven har som tidligere nevnt, ikke egne bestemmelser om konsesjon (forhåndstillatelse) til å behandle helseopplysninger, men viser i § 5 til personopplysningsloven § 33. I § 5 annet ledd sies det klart at helseregistre som skjer med hjemmel i forskrift etter helseregisterloven §§ 6 – 8, ikke krever konsesjon. Det betyr at verken behandlingsrettede helseregistre (§ 6) eller lokale, regionale og sentrale helseregistre (§§ 7 og 8), trenger søke konsesjon, fordi lovgiver og Kongen har tatt stilling til spørsmålene i helseregisterloven med tilhørende forskrifter. All annen behandling av helseopplysninger må i utgangspunktet ha hjemmel i lov, fordi helseopplysninger er definert som ”sensitive” i personopplysningsloven § 2 nr 8 bokstav c, og konsesjonsplikten i personopplysningsloven § 33 utløses av det faktum at det blir behandlet helseopplysninger. Alle forskningsprosjekter som innebærer behandling av én eller flere opplysningstyper som er sensitive (dvs som gjelder helseforhold og / eller andre typer innhold som er definert som sensitivt i personopplysningsloven § 2 nr 8), må i utgangspunktet ha forhåndstillatelse (konsesjon) fra Datatilsynet. I personopplysningsforskriften § 7-27 stilles det opp et generelt unntak fra konsesjonsplikt for sensitive opplysninger som behandles for forskningsformål:
”Førstegangs kontakt opprettes på grunnlag av offentlig tilgjengelig informasjon eller gjennom en faglig ansvarlig person ved virksomheten der respondenten er registrert, eller respondenten selv tar kontakt med prosjektleder eller dennes representant,
respondenten har samtykket i alle deler av undersøkelsen. Dersom respondenten er umyndig, kan en annen med samtykkekompetanse for respondenten avgi slikt samtykke,
prosjektet skal avsluttes på et tidspunkt som er fastsatt før prosjektet settes i gang,
det innsamlede materialet anonymiseres eller slettes ved prosjektavslutning, og
prosjektet ikke gjør bruk av elektronisk sammenstilling av personregistre.”
Vilkårene er kumulative, og innebærer at et stort antall medisinske forskningsprosjekter ikke omfattes av unntaket, men er avhengig av konsesjon fra Datatilsynet.
Selve konsesjonsbehandlingen innebærer en avveining mellom hensyn som ofte fremstår som inkommensurable. På den ene siden skal det legges vekt på hensynet til personvern, jf avsnitt 14.2, og på den annen side skal det legges vekt på de hensyn som taler for at databehandlingen utføres, for eksempel hensynet til å vinne ny medisinsk kunnskap for å forebygge og behandle sykdom mv. Også utsiktene til økonomiske / industriell utnyttelse av forskningsresultater og offentlig behov for styring av helsetjenestene mv, er relevante for vurdering av om konsesjon skal gis. Mens hensynene på ”personvernsiden” i hovedtrekk er vel definerte og velordnede, er de hensyn som taler for gjennomføring av helseforskning relativt flere og mer ”sprikende”. Datatilsynet kan i henhold til personopplysningsloven § 35 stille vilkår for at konsesjon skal bli gitt. I praksis vil det kunne oppstå en forhandlingslignende situasjon, der Datatilsynet og konsesjonssøker prøver å finne frem til ordninger som kan forene motstridende interesser og på den måten unngå at søknaden avslås.
Dersom det ikke foreligger konsesjonsplikt, fordi behandlingen kommer inn under forskriftene til helseregisterloven §§ 6 – 8 eller unntaket i personopplysningsforskriften § 7-27, skal behandlingen i utgangspunktet meldes til Datatilsynet. Fordi Norges samfunnsvitenskapelige datatjeneste (NSD) er utpekt som personvernombud for forskning ved universiteter og høyskoler, 89 skal melding om forskningsprosjekter ved disse institusjonene skje til NSD. Forskingsinstitusjoner utenfor universitets- og høgskolesektoren (som ikke er knyttet til denne ordningen), må sende melding til Datatilsynet. Meldingen skal inneholde ni ulike opplysningstyper, se helseregisterloven § 30 første ledd. Blant disse er formålet med behandlingen, hvilke typer helseopplysninger som planlegges behandlet, hvilke kilder som ønskes anvendt og hvor opplysningene eventuelt skal utleveres. Dersom det senere blir endringer i noen av de opplysninger det er sendt melding om, skal ny melding sendes.
14.4 Særregulering av visse helseregistre
Helseregisterloven inneholder en rekke bestemmelser som gir Kongen kompetanse til å forskriftsregulere behandlingsrettede helseregistre (jf § 6), regionale og lokale helseregistre (jf § 7) og sentrale helseregistre (jf § 8). I medhold av helseregisterloven §§ 6 og 8 er det vedtatt forskrifter for i alt åtte slike registre:
Forskrift om innsamling og behandling av helseopplysninger i Reseptbasert legemiddelregister (Reseptregisteret) 90
Forskrift om tuberkulosekontroll 91
Forskrift om innsamling og behandling av helseopplysninger i Meldingssystem for smittsomme sykdommer og i Tuberkuloseregisteret og om varsling om smittsomme sykdommer (MSIS- og Tuberkuloseregisterforskriften) 92
Forskrift om innsamling og behandling av helseopplysninger i Dødsårsaksregisteret (Dødsårsaksregisterforskriften). 93
Forskrift om innsamling og behandling av helseopplysninger i Kreftregisteret (Kreftregisterforskriften). 94
Forskrift om innsamling og behandling av helseopplysninger i Medisinsk fødselsregister (Medisinsk fødselsregisterforskriften) 95 .
Forskrift om innsamling og behandling av helseopplysninger i System for vaksinasjonskontroll (SYSVAK-registerforskriften). 96
Forskrift om innsamling og behandling av helseopplysninger i Norsk overvåkingssystem for antibiotikaresistens hos mikrober (NORM-registerforskriften). 97
Her skal vi ikke gå inn i detaljer på forskriftene, men i stedet gjøre en overordnet analyse av forskriftenes betydning i relasjon til medisinsk og helsefaglig forskning.
Sju av åtte helseregisterforskrifter 98 følger stort sett samme standardstruktur, med en kapittelinndeling som også angir de viktigste problemstillingene som forskriftene regulerer. Alle sju forskrifter har således et innledningskapittel der følgende spørsmål er nærmere behandlet: 99
Formålet med forskriften.
Hvem som er databehandlingsansvarlig (”databehandlingsansvarlig”) for vedkommende register.
Hvem som kan være databehandler for vedkommende register.
Hvilke opplysningstyper som kan inngå i vedkommende register.
Angivelse av hva som er lovlig eller ulovlig anvendelse av vedkommende register.
Alle forskrifter har formålsbestemmelser og i alle forskrifter, unntatt forskriften om tuberkulosekontroll, er forskning ett av flere formål med bruken av registrene som eksplisitt er angitt i loven. 100 Forskriften om tuberkulosekontroll har en generell formålsbestemmelse i § 1-1 som i stor grad er formulert med tanke på tiltak mot tuberkulose, og der forskning verken er nevnt i forskriften selv eller i departementets kommentarer til forskriften.
Forskriftene gir Norsk folkehelseinstitutt databehandlingsansvaret for alle de nevnte registrene, med unntak av Kreftregisteret (som institusjonen Kreftregisteret har databehandlingsansvaret for). I seks av sju tilfeller har Folkehelseinstituttet hjemmel til å inngå skriftlige avtaler med databehandlere uten særskilte begrensninger. I NORM-registerforskriften er det imidlertid fastsatt at Universitetssykehuset i Nord-Norge skal ha databehandlingsansvar, og andre databehandlere kan derfor ikke velges.
Fem av registerforskriftene har bestemmelser om ”Forbud mot bruk” (§ 1-4) som knytter an mot formålet med registeret. Således bestemmes det i disse forskriftene at opplysningene ”ikke anvendes til formål som er uforenelig med formål som nevnt [...]” i formålsbestemmelsen. I reseptregisterforskriften heter det at det bare er lovlig å bruke opplysninger i tråd med formålet, mens det i dødsårsaksregisterforskriften ikke finnes en bestemmelse om lovlig / ulovlig bruk. De nevnte vilkårene om at bruken av opplysninger ikke skal være uforenlig med formålet, står i kontrast med kravet til fastsettelse av formål i helseregisterloven, se særlig § 8 fjerde ledd for de sentrale helseregistrene. Forskriftsbestemmelsene må derfor trolig tolkes innskrenkende slik at all bruk av opplysningene må være i tråd med det definerte formålet.
Forskriftene regulerer adgangen til å sammenstille opplysninger fra vedkommende register med andre helseopplysninger. Slik sammenstilling kan normalt bare skje i forhold til andre konkrete registre som er eksplisitt nevnt i forskriften, og dersom resultatet av sammenstillingen fremstår som anonym. For enkelte registre er det også krav til at sammenstillingen ikke skal være etisk betenkelig, og det kan være stilt krav om hvem som skal utføre sammenstillingen.
Utlevering av opplysninger fra helseregistrene, dvs overføring til mottakere utenfor databehandlingsansvarliges eller databehandleres organisasjon, kan normalt bare skje i form av statistikk og avidentifiserte opplysninger. Det kreves dessuten at utlevering skal være etisk ubetenkelig, og kan stilles krav til hvor tilrettelegging og behandling av utleverte opplysninger kan skje.
Når det gjelder sammenstilling og utlevering av helseopplysninger i sentrale registre, innebærer forskriftsreguleringene at den adgangen som forskriftene fastsetter er ”garantert” og ikke avhengig av konsesjon fra Datatilsynet. Adgangen til ytterligere sammenstilling og utlevering er ikke stengt, men avhengig av konsesjon etter personopplysningsloven § 33, jf. helseregisterloven § 12. 101
Sju av åtte helseregisterforskrifter har bestemmelser om ansvar for og kompetanse til å fastsette og dokumentasjon av kodeverk. Primæransvaret for dette ligger hos den databehandlingsansvarlige, mens departementet som oftest har kompetanse til å gi nærmere bestemmelser om kodeverk, og kan herunder fastsette koder i henhold til nasjonale og internasjonale normer. I NORM-registerforskriften er det dessuten fastsatt at databehandlingsansvarlige skal sørge for at de opplysninger som behandles i NORM er korrekte, relevante og nødvendige for de formål de samles inn for. Konkret vil dette bl.a. innebære at Nasjonalt folkehelseinstitutt (som databehandlingsansvarlig) er ansvarlig for at opplysningene har den kvalitet som kreves for å gjennomføre forskning. I dette ligger det imidlertid ikke annet enn hva som følger av de generelle bestemmelsene i helseregisterloven § 11 første ledd. Alle forskrifter må således forstås slik at de institusjoner som har databehandlingsansvaret, skal sørge for at opplysningene har den kvalitet som kreves ut i fra de formål som er angitt i forskriftene, herunder for forskningsformål. I tillegg må imidlertid opplysningskvaliteten vurderes ut i fra de krav som følger av det konkrete forskningsprosjekt. Dersom opplysningene utleveres til andre 102 , får mottaker av opplysningene (for eksempel en forskningsinstitusjon) et selvstendig databehandlingsansvar, som bl.a innebærer en plikt til å sikre opplysningskvaliteten.
Den videre kapittelstrukturen i alle de nevnte forskrifter, unntatt forskriften om tuberkulosekontroll, følger stort sett dette mønster der en gir bestemmelser om:
melding til registeret,
den nærmere behandling av opplysninger i registeret,
taushetsplikt
innsyn for den enkelte
bevaring og langtidslagring av opplysningene i registeret.
Enkelte forskrifter har kapitler med særlige bestemmelser, for eksempel har MSIS- og tuberkuloseregisterforskriften et eget kapittel om varsling av smittsomme sykdommer. I tillegg er det i alle forskrifter gitt bestemmelser om straff for brudd på forskriftsbestemmelsene og ikrafttredelse av forskriften mv.
Med unntak av opplysningene i Resept- og NORM-registeret har registrerte personer normalt rett til informasjon og innsyn i tråd med bestemmelsene i helseregisterloven §§ 22 – 25. De to registrene der det ikke er særskilt regulering av retten til innsyn og informasjon inneholder ikke direkte identifiserbare opplysninger (men henholdsvis pseudonymiserte og avidentifiserte helseopplysninger). For alle registre gjelder i tillegg reglene om generell informasjon i henhold til helseregisterloven §§ 20 og 21.
Hovedregelen for de forskriftsregulerte helseregistrene er at opplysningene kan oppbevares på ubestemt tid. Unntak kan gjelde for mangelfulle opplysninger (helseregisterloven § 26) og opplysninger som er belastende for den registrerte (helseregisterloven § 28).
14.5 Avsluttende bemerkninger
I dette kapittelet er det gjort et forsøk på å gi en ordnet fremstilling av helseregisterloven, samtidig som det er gitt et sideblikk til personopplysningsloven. Fremstillingen viser at vi her står overfor to kompliserte regelverk som begge kan få anvendelse på medisinsk- og helsefaglig forskning. Begge lover omfatter flere bestemmelser og har større detaljeringsgrad enn hva det trolig er behov for innen medisinsk- og helsefaglig forskning. Begge lover er dessuten mangelfulle på enkelte felt som har stor betydning for slik forskning, særlig med hensyn til autonomien til deltakere i forskningsprosjekter. Det er på denne bakgrunn grunn til å undersøke mulighetene for å erstatte begge lover med egne lovbestemmelser som særlig gjelder behandling av helseopplysninger i forskningssammenheng. Se nærmere om utvalgets vurderinger av en slik særregulering i kapittel 30.
15 Åndsretten m.m.
15.1 Innledning
Åndsretten eller immaterialretten omfatter ”hovedsakelig rettsregler som gir enerett til resultatene av intellektuell og skapende virksomhet, og til kjennetegn.” 103
Åndsretten er relevant i denne sammenheng ved at den omhandler forskernes rettigheter. Åndsretten hører hjemme i det jurister ofte kaller privatrett, dvs. regler som regulerer rettsforholdet mellom private individer. Privatrettslige regler er på mange måter nokså forskjellige fra de offentligrettslige reglene beskrevet foran, og som først og fremst regulerer forholdet mellom individ og samfunn.
I dette avsnittet omtales patentloven og åndsverksloven kort. I tillegg omtales arbeidstakeroppfinnelsesloven som særlig omhandler forholdet mellom arbeidstaker (oppfinner) og arbeidsgiver.
Det grunnleggende formålet med åndsretten er å stimulere til utvikling og nyvinninger gjennom belønninger til oppfinnere i form av eneretter.
15.2 Patentloven
I hvilken utstrekning det er anledning til å oppnå patent på blant annet forskningsresultater, reguleres av patentloven. Det å inneha patent, innebærer at patentinnehaveren er gitt en tidsbegrenset enerett til kommersiell utnyttelse av en oppfinnelse, jf. patentloven § 1 første ledd sammenholdt med § 40.
Patentrettslig enerett omfatter alle former for kommersiell utnyttelse i næringsvirksomhet, for eksempel produksjon, markedsføring, omsetting og bruk. Patentbeskyttelsen innebærer at andre interessenter ikke kan benytte oppfinnelsen kommersielt uten patentinnehaverens tillatelse, jf. § 3 første og andre ledd. Slik tillatelse vil ofte kunne være betinget av økonomisk godtgjørelse. Patent gir imidlertid ikke patentinnehaveren noen automatisk rett til å utnytte oppfinnelsen. Bruken kan for det første være forbudt etter annen lovgivning, og andre patentrettigheter kan hindre utnyttelse av det aktuelle patent.
I forbudet mot kommersiell utnyttelse uten tillatelse er det gjort enkelte unntak i § 3 tredje ledd. Det fremgår her at utnyttelse som ikke skjer i nærings- eller driftsøyemed, ikke trenger tillatelse. Likeledes dersom utnyttelsen skjer ved et eksperiment som angår selve oppfinnelsen. Det vil således være adgang til å forske på en patentert oppfinnelse.
Som nevnt må det ha funnet sted en oppfinnelse før det kan gis patent. Oppfinnelsen kan bestå i en anvendelsesmetode, en fremgangsmåte eller et produkt, for eksempel kjemiske produkter eller tekniske apparater, eller en kombinasjon av disse. I tillegg må oppfinnelsen ha teknisk karakter eller ha teknisk effekt slik at den løser et problem, og oppfinnelsen må være reproduserbar. På bakgrunn av den beskrivelse som er gjort av oppfinnelsen i patentsøknaden, skal andre med relevant fagkunnskap kunne ”gjenta” oppfinnelsen, jf. § 8 andre ledd.
For at det skal foreligge en oppfinnelse i lovens forstand, må oppfinnelsen være:
Ny og ikke tidligere kjent, jf. § 2 første ledd.
Skille seg vesentlig fra kjent teknikk, jf. § 2 første ledd.
Industrielt anvendbar, jf. § 1 første ledd.
Grensen mellom oppfinnelser (som kan patenteres) og oppdagelser (som ikke kan patenteres) blir etter dette viktig. Det å erkjenne at et nytt materiale finnes i naturen, eller å erkjenne en ny egenskap hos et tidligere kjent materiale, vil kun representere en oppdagelse. Derimot vil utnyttelsen av en slik erkjennelse kunne utgjøre en oppfinnelse i patentlovens forstand. Dette kan for eksempel være tilfellet etter at materialet er isolert, bearbeidet eller modifisert slik at det dermed kan løse et teknisk problem. Ved helhetsvurderingen av om det kan sies å foreligge en oppfinnelse, vil det imidlertid være adgang til å legge en viss vekt på det sprang i teknisk utvikling som selve oppdagelsen av et naturlig forekommende materiale innebærer.
Av § 1 fjerde ledd, fremgår det at det ikke er adgang til å oppnå patent på oppfinnelser hvis utnyttelse ville stride mot sedelighet eller offentlig orden. Videre fremgår av § 1 fjerde ledd at det ikke er adgang til å oppnå patent på ”plantesorter eller dyreraser eller vesentlige biologiske fremgangsmåter for fremstilling av planter eller dyr”. Dette omfatter alle planter og dyr, herunder encellede planter og dyr, samt differensierende celler. Likedan vil det ikke være adgang til å oppnå patent på fremgangsmåten til fremstilling av slike.
Unntakene i § 1 fjerde ledd, omfatter i utgangspunktet ikke humant biologisk materiale. Som følge av grunnvilkårene om at det skal foreligge en nyhet som skiller seg fra det som allerede var allment kjent, samt at det må beskrives en industriell anvendbarhet, vil det imidlertid ikke kunne oppnås patent på mennesker, fostre, organer eller vev som sådan, befruktede og ubefruktede egg eller sædceller. Det er med andre ord ikke adgang til å oppnå patent på humant biologisk materiale slik det foreligger naturlig i mennesket. Det er imidlertid mulig å oppnå patent på for eksempel kjemiske produkter eller teknisk utstyr som er nødvendig for å påvise humant biologisk materiale som foreligger i sin naturlige form, eller påvise naturlige prosesser som skjer i humant biologisk materiale. Som eksempel vil man ikke kunne patentere en human celle, men man vil kunne patentere teknisk utstyr som gjør det mulig å studere cellen. Dette forutsetter imidlertid at slikt teknisk utstyr virkelig er nytt, at det er anvendbart og at det skiller seg vesentlig fra tidligere allment kjent utstyr.
Når det gjelder isolerte og bearbeidede organer og vev, vil det her kunne oppnås patent, men bare dersom også grunnvilkårene er oppfylt. Likedan vil det kunne oppnås patent på bearbeidede celler eller gener. Når det særlig gjelder patent på gener, skal det bemerkes at gener bare kan patenteres dersom man vet hva genet koder for, eller hvilken funksjon det har. Dette innebærer ikke patent på genet slik det foreligger i det menneskelige legeme men patent på genet slik dette uttrykker sin funksjon. Det vil neppe være et krav for å få patent at man har beskrevet alle funksjoner et gen kan tenkes å ha. I så måte kan det gis flere patenter på samme gen dersom man oppfinner en ny funksjon eller anvendelse. På bakgrunn av forskning utført på humant biologisk materiale vil man kunne patentere de oppfinnelser man gjør, og deretter benytte dette i næringsutvikling eller industriell sammenheng dersom man sikrer seg patent på produkter som er utviklet etter genstudier eller studier av geners funksjon.
15.3 Åndsverksloven
Åndsverkloven fastsetter de nærmere rettsregler om beskyttelsen og utnyttelsen av åndsverk. Med åndsverk forståes i denne lov litterære, vitenskapelige eller kunstneriske verk av enhver art og uansett uttrykksmåte og uttrykksform, jf. § 1 annet ledd. Loven gjelder uavhengig av hvilken måte og form åndsverket har kommet til uttrykk på.
Den som skaper et åndsverk, har opphavsrett til verket, jf. § 1 første ledd. Hovedregelen er at den enkelte opphavspersonen til et åndsverk har rett til å bestemme om og hvordan åndsverket skal gjøres tilgjengelig for andre. Opphavspersonen gis en enerett til å råde over åndsverket ”ved å fremstille eksemplarer av det og ved å gjøre det tilgjengelig for allmennheten, i opprinnelig eller endret skikkelse, i oversettelse eller bearbeidelse, i annen litteratur- eller kunstart eller i annen teknikk” jf. åndsverkloven § 2. Man trenger ikke å søke om det eller på annen måte registrere en slik rett.
I praksis betyr dette at det ikke kan disponeres over et åndsverk uten opphavspersonens samtykke. Dette sikrer at opphavspersonen ikke er rettsløs etter at vedkommendes åndsverk er blitt kjent for allmennheten i en eller annen form som kan utnyttes til eksemplarfremstilling (kopiering), spredning av eksemplar eller offentlig fremføring. Rettsreglene sikrer at opphavspersonen kan regulere slik bruk av sitt verk og gir vedkommende derved også mulighet for et økonomisk utkomme av det han eller hun har skapt.
15.4 Arbeidstakeroppfinnelsesloven og kommersialisering av forskningsresultater
I følge arbeidstakeroppfinnelsesloven har universiteter og høgskoler, dersom de ønsker det, anledning til å overta retten til næringsmessig utnyttelse av patenterbare oppfinnelser gjort av ansatte ved institusjonen. For å sikre forskerne mulighet for åpen kommunikasjon av kunnskap, er det gjort unntak fra lovens generelle bestemmelser. Unntaket innebærer at lærere og vitenskapelig personale i hel- eller deltidsstillinger ved universiteter og høgskoler, har en lovfestet rett til fritt å publisere sine forskningsresultater, selv når dette kan hindre institusjonen i å utnytte en oppfinnelse næringsmessig. Publiseringsretten må være benyttet innen ett år etter at institusjonen er varslet om oppfinnelsen, for at institusjonen ikke skal kunne overta retten til oppfinnelsen (lovens § 6, tredje ledd).
Forskere må varsle institusjonen uten ugrunnet opphold dersom det foreligger et forskningsresultat eller en oppfinnelse som kan antas å være patenterbar. Eventuelle verdier fra oppfinnelser fordeles mellom forsker og institusjon. Ved ekstern finansiering av forskningsprosjekter, må rettighetsforholdet avklares gjennom separate avtaler mellom institusjon, finansieringskilde og forsker, forut for prosjektets oppstart.
Ifølge Undervisnings- og forskningsdepartementet 104 gir loven en rimelig balansering av rettigheter og plikter for forsker og institusjon, og gjør det praktisk lettere å organisere arbeidet med næringsmessig utnyttelse av oppfinnelser ved universiteter og høgskoler. Forskerne gis hjelp av institusjonen i arbeidet med utnyttelsen av oppfinnelsen, og sikrer dem anledning til å konsentrere seg om forskning og undervisning. Institusjonen må på sin side organisere et apparat som kan sikre forskernes og institusjonens rettigheter, og som sørger for at oppfinnelser blir patentert og formidlet til næringslivet.
16 Erstatningsrett
16.1 Alminnelig erstatningsrett
I henhold til alminnelige erstatningsrettslige regler kan forskeren eller den institusjon han eller hun arbeider ved, bli holdt ansvarlig for handlinger som medfører skade og/eller økonomisk tap. Etter alminnelige erstatningsrettslige prinsipper må tre hovedvilkår være oppfylt for at erstatning skal kunne tilkjennes:
Det må foreligge et ansvarsgrunnlag, for eksempel uaktsomhetsansvar (culpa) eller objektivt ansvar.
Det må foreligge en skade som materialiserer seg i et økonomisk tap.
Det må være årsakssammenheng mellom ansvarsgrunnlaget og skaden/tapet.
Erstatningsretten har i mange land, særlig i USA, spilt en sentral rolle ved identifiseringen av ulovfestede rettsregler vedrørende medisinsk forskning, blant annet ved at pasienter og forsøkssubjekter har anlagt erstatningssaker mot leger og forskere, jf. kapittel 9.
Den alminnelige erstatningsrett er i hovedsak ulovfestet og kommer til anvendelse på alle livsområder. I tillegg finnes det særskilte erstatningsregler i spesiallovgivning, som gjelder på bestemte områder. Pasientskadeloven og produktansvarsloven er her eksempler. Det vises til kapittel 33 punkt kapittel 31.2.2 for en inngående drøftelse av de erstatningsrettslige spørsmål som medisinsk og helsefaglig forskning reiser.
16.2 Pasientskadeloven
Pasientskadeloven av 2001 tar sikte på å styrke pasientens erstatningsrettslige stilling. Loven gjelder etter en lovendring også medisinsk forsøksvirksomhet, jf. § 1, 2. ledd. I følge § 1 gjelder loven ”skader som er voldt
i institusjon under spesialisthelsetjenesten og kommunehelsetjenesten,
under ambulansetransport, eller
av helsepersonell som yter helsehjelp i henhold til offentlig autorisasjon eller lisens, personer som opptrer på vegne av disse eller andre personer som fastsatt i forskrift.
Slike skader regnes som pasientskader dersom de er voldt under veiledning, undersøkelse, diagnostisering, behandling, ekspedisjon av legemidler fra apotek, pleie, vaksinasjon, prøvetaking, analyse av prøver, røntgen, forebygging av helseskader, medisinsk forsøksvirksomhet samt donasjon av organer, blod og vev.”
I § 2, 1. ledd heter det at pasienten og andre som har lidt tap på grunn av pasientskade, har krav på erstatning når skaden skyldes
svikt ved ytelsen av helsehjelp, selv om ingen kan lastes,
teknisk svikt ved apparat, redskap eller annet utstyr som er brukt ved ytelsen av helsehjelp,
smitte eller infeksjon, når dette ikke i hovedsak skyldes pasientens tilstand eller sykdom, vaksinasjon, eller
forhold som medfører ansvar for helsetjenesten eller helsepersonell etter alminnelige erstatningsregler.
Det skal tas hensyn til om de krav skadelidte med rimelighet kan stille til virksomheten eller tjenesten på skadetidspunktet, er tilsidesatt. Påregnelige følger av behandling gis det ikke erstatning for. Loven presiserer også at utilstrekkelige ressurser ikke skal medføre ansvar dersom ressursfordelingen har vært forsvarlig og virksomheten i alminnelighet holder en forsvarlig standard.
Selv om det ikke foreligger grunnlag for erstatningsansvar etter første og annet ledd, kan det unntaksvis ytes erstatning når det har skjedd en pasientskade som er særlig stor eller særlig uventet, og som ikke kan anses som utslag av en risiko som pasienten må akseptere. Det skal legges vekt på om det er gitt tilstrekkelig informasjon på forhånd.
Loven stiller ikke krav om at skaden må manifestere seg på sykehuset. Det er dermed tilstrekkelig, som ordlyden også tilsier, at skaden er voldt under forsøket. Hvor skaden blir merkbar eller synlig er uten betydning. I denne sammenheng kan nevnes at det ikke bare er fysiske skader som kan være pasientskader, men også psykiske skader.
Ved avgjørelsen av om erstatning skal tilkjennes, skal det tas hensyn til om de krav skadelidte med rimelighet kan stille til virksomheten eller tjenesten på skadetidspunktet, er tilsidesatt, jf § 2 annet ledd. Ved medisinsk forskning vil vurderingen av hva skadelidte med rimelighet kunne forvente, også måtte avgjøres på bakgrunn av de alminnelige prinsipper og regler som gjelder for medisinsk forsøksvirksomhet.
Ansvarsgrunnlaget er, som det fremgår av ordlyden, i de fleste tilfeller betinget av at skaden skyldes en form for feil, en svikt. Selv om ingen kan bebreides, er det et krav at noe har gått galt for at erstatning skal innrømmes. Erstatning gis bare når det blir krevd, og kravet skal meldes skriftlig til Norsk Pasientskadeerstatning, som er ansvarlig for skade som kan kreves erstattet etter loven. Hvis skadelidte ikke er enig i avgjørelsen om erstatning, kan avgjørelsen klages inn for Pasientskadenemda eller prøves for domstolene.
Se for øvrig punkt 33.2.3 for en nærmere drøftelse av disse spørsmål.
16.3 Produktansvarsloven
Produktansvarsloven har bakgrunn i alminnelige ulovfestede erstatningsregler, kjøpsrettslige mangelsbetraktninger og behovet for forbrukervern.
Produktansvarsloven innfører en tvungen forsikringsordning - Legemiddelforsikringen - som gjelder for alle importører og produsenter av legemidler, for å dekke personskade voldt av legemiddel.
Lovens kapittel 3 fastsettes særregler om legemiddelansvaret. Det er et objektivt ansvar, uten hensyn til skyld eller om legemidlet byr på den sikkerhet som en bruker med rimelighet kunne forvente eller ikke. Skadelidte har i utgangspunktet krav på erstatning fra Legemiddelforsikringen så fremt legemidlet volder skade.
Det vises til punkt 33.2.4 for en nærmere drøftelse av disse spørsmål.
16.4 Ansvarsforsikring
Det eksisterer ingen generell forsikringsplikt for forskere, slik det for eksempel gjør for leger. Unntak gjelder ved klinisk utprøving av legemidler, jf. ovenfor om produktansvarsloven.
Det er tvilsomt om den alminnelige ansvarsforsikringen som leger har, også dekker forskningsvirksomhet som det ikke er naturlig å se på som legevirksomhet, for eksempel ikke-terapeutisk forskning. De fleste forskere er også helsepersonell og arbeider i helsetjenesten, og deres virksomhet (også forskningsvirksomhet) vil dermed kunne være dekket av pasientskadeloven og Norsk Pasientskadeerstatning, hvis de er en del av offentlig helsetjeneste inklusive fastlegeordningen, men ikke som helt private aktører. På samme måte vil legemiddelindustrien vil være dekket gjennom Legemiddelforsikringen, jf. produktansvarsloven. Se kapittel 25 og kapittel 33.
17 Strafferett
Formålet med strafferettslige forbuds- og sanksjonsregler er å hindre forekomst av uønskede handlinger. Helsearbeidere og forskere kan møtes med straffereaksjoner i den grad overtredelse av lovgivningen er belagt med straff. Noen av de første rettslige reguleringene av medisinsk forskning har opphav i den alminnelige strafferett, jf. kapittel 9.
Det rettslige utgangspunkt etter norsk rett, er at handlinger er tillatt med mindre de er forbudt i medhold av lovgivning eller annet tilstrekkelig rettsgrunnlag. Straffeloven inneholder ingen bestemmelser som spesifikt har til hensikt å hindre visse typer medisinsk og helsefaglig forskning. Imidlertid vil man ofte ved medisinsk forskning foreta kroppslige inngrep. Slike inngrep vil som utgangspunkt måtte bli å betrakte som en legemskrenkelse som omfattes av straffelovens forbud mot slike, jf. straffelovens §§ 228 flg. Et samtykke fra personen selv eller den som har samtykkekompetanse på hans eller hennes vegne, eventuelt andre straffriende forhold, vil imidlertid gjøre den ellers straffbare legemskrenkelsen både rettmessig og straffri, jf. straffelovens § 235, 1. ledd.
Straffelovens § 222 oppstiller forbud mot tvang, det vil si ved rettsstridig atferd eller ved å true med sådan tvinge noen til å gjøre, tåle eller unnlate noe. Forskere kan ikke ved hjelp av trusler eller annen rettsstridig atferd tvinge noen til å delta i forsk-ning. Bestemmelsene i straffeloven springer ut av oppfatningen om at individets legeme og frihet er undergitt et strengt rettslig vern, og at individet har rett til å motsette seg uønskede inngrep i egen kropp og har rett til fravær av trusler og tvang. Dette gjelder også ved alle former for medisinsk og helsefaglig forskning.
18 Veiledning, forhåndsvurdering og tilsyn
18.1 Innledning
I dette kapittelet beskrives de organer som har veilednings-, kontroll- og/eller tilsynsoppgaver i forhold til dagens regelverk vedrørende medisinsk og helsefaglig forskning.
Den rettslige reguleringen av forskning som er beskrevet ovenfor, skal sikre at forskere utøver sin virksomhet forsvarlig og opptrer i overensstemmelse med vedtatte normer. Reguleringene må følges opp med informasjon, veiledning og holdningsskapende virksomhet. I tillegg må det føres kontroll og tilsyn med den aktuelle virksomhet for å sikre at de rettslige regler blir forstått og etterfulgt.
Forskere er ansvarlige for å utøve sin virksomhet i tråd med gjeldende reguleringer (samfunnets aksepterte atferdsnormer). Det at vedkommende vet at han/hun vil kunne bli kontrollert og stilt til ansvar dersom han/hun ikke opptrer innenfor gjeldende normer og regler, antas å ha en preventiv effekt mot regelbrudd.
Ulike former for kontroll og tilsyn
Det er tre hovedformer for rettslig kontroll og tilsyn:
Forhåndskontroll (før prosjektet settes i gang)
Løpende kontroll og tilsyn (mens prosjektet gjennomføres)
Etterhåndskontroll (etter at prosjektet (eksperimentet) er avsluttet).
Gjeldende kontroll- og tilsynssystem i Norge er hovedsakelig konsentrert omkring forhåndskontroll. Det innebærer at de fleste medisinske forskningsprosjekter som involverer mennesker, må ha blitt tilrådd, godkjent, fått konsesjon eller lignende før de kan settes i gang. Det er lagt til grunn at det av hensyn til forskningsdeltakerne og samfunnet for øvrig, er nødvendig å kontrollere at medisinske forsøk er planlagt utført i overensstemmelse med gjeldende lover og regler. Man vil med andre ord sikre at prosjektene er faglig og etisk forsvarlige. Det er da også i denne fasen det er mest aktuelt med veiledning og rådgivning. I dag bruker De regionale komiteene for medisinsk forskningsetikk (REK) mye tid og ressurser på å veilede forskere når det gjelder de etiske sidene ved et prosjekt og utvikle prosjekter i dialog med forskerne. Det samme gjelder andre forvaltningsorganer, som for eksempel Statens legemiddelverk, i henhold til den veiledningsplikten som følger forvaltningsloven § 11.
Selv om prosjektet er godt planlagt, er det imidlertid ikke sikkert at selve utøvelsen forløper som planlagt. Enkelte organer fører av den grunn løpende kontroll eller tilsyn på visse områder.
Til tross for den omfattende forhåndskontrollen føres det i dag ingen oversikt over avsluttede prosjekter (etterhåndskontroll). Man vet derfor ikke hvordan det går med alle de prosjektene som er tilrådd eller godkjent. Legemiddelforskriften representerer her et unntak, ved at § 5-9 etablerer en form for etterhåndskontroll ved klinisk utprøving av legemidler (se nedenfor). Komitésystemet planlegger også et register. Etterhåndskontroll kan også skje dersom det i ettertid klages over forsøket, for eksempel fra forsøkspersoner som mener de har lidd overlast.
Ulike typer kontroll- og tilsynsorganer
Kontroll- og tilsynsorganene kan deles inn i tre hovedkategorier:
Offentlige myndighetsorganer, for eksempel Helse- og omsorgsdepartementet og Datatilsynet. Dette er organer som med hjemmel i lov blant annet driver med offentlig myndighetsutøvelse, herunder kontroll og/eller tilsyn med de deler av medisinsk forskningsvirksomhet som faller innenfor deres myndighetsområde. Alle offentlige forvaltningsorganer er underlagt forvaltningsloven. Loven stiller opp en del sentrale saksbehandlingsregler som skal ivareta borgerens rettsikkerhet i møte med det offentlige. 105 Dette er grunnleggende regler om god forvaltningsskikk, habilitet, taushetsplikt, begrunnelse av vedtak og klageadgang. Dessuten er alle forvaltningsorganer pålagt å gi råd gjennom den alminnelige veiledningsplikten. Offentlighetsloven skal sikre offentligheten innsyn i forvaltningens virksomhet. Innsynsretten gjør det mulig å føre kontroll med kontrollørene. Domstolene er underlagt tilsvarende prosessuelle regler.
Myndighetsnære organer, for eksempel de regionale komiteer for medisinsk forskningsetikk (REK). Dette er organer som formelt sett ikke driver myndighetsutøvelse, men som på grunn av sin karakter og kontrollfunksjon ligger så nær slik virksomhet at det er naturlig å sammenligne dem med offentlige myndighetsorganer.
Frie aktører, for eksempel Norsk Samfunnsvitenskapelig Datatjeneste AS (NSD), institusjonsbaserte forskningsutvalg og lignende. Dette er en uensartet gruppe organer, gjerne opprettet av private aktører som ikke har som primærformål å drive med myndighetsutøvelse, men heller kvalitetssikre forskning som de selv på ulike måter er involvert i.
Det eksisterende systemet oppleves av mange forskere som uoversiktlig, noe som synliggjøres av figur 18.1.
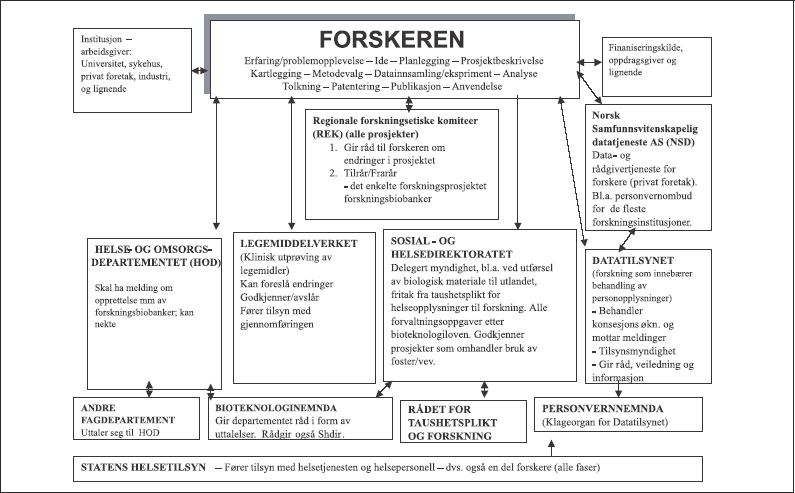
Figur 18.1 Finansiering av medisinsk og helsefaglig forskning i universitets- og høyskolesektoren i 2001. I tillegg kommer instituttsektoren og den andelen medisinsk og helsefaglig forskning som næringslivet utfører i egen regi.
18.2 Offentlige myndighetsorganer
18.2.1 Helse- og omsorgsdepartementet
Helse- og omsorgsdepartementet 106 er overordnet offentlig myndighet på helseområdet. Departementet har et faglig og administrativt etatsstyringsansvar for Sosial- og helsedirektoratet, Statens helsetilsyn, Nasjonalt folkehelseinstitutt, Statens legemiddelverk, Statens strålevern, Bioteknologin-emnda, og Medinnova SF. Departementet har i tillegg oppgaver knyttet til eierstyringen av de fem regionale helseforetakene. Helse- og omsorgsdepartementet har også ansvaret for enkelte oppgaver i Trygdeetaten og Mattilsynet.
De mer spesifikke forvaltningsoppgaver vedrørende medisinsk og helsefaglig forskning er delegert til underliggende etater.
Helse- og omsorgsdepartementet skal imidlertid ha melding om opprettelse av forskningsbiobanker i henhold til biobankloven § 4. Opprettelse av biobanker skal først forelegges en regional komité for forskningsetikk (REK). Departementet kan nekte opprettelse dersom etiske hensyn eller tungveiende samfunnsmessige interesser tilsier det. Departementet fører et register over innmeldte biobanker, jf. § 6.
18.2.2 Sosial- og helsedirektoratet
Sosial- og helsedirektoratet (direktoratet) 107 er et statlig forvaltningsorgan underlagt Helse- og omsorgsdepartementet som skal bidra til å gjennomføre og iverksette nasjonal politikk i helse- og sosialsektoren. Direktoratet ble etablert 1. januar 2002 etter en sammenslåing av en rekke offentlige organer. Direktoratet har fått delegert mange forvaltningsoppgaver knyttet til medisinsk forskning fra Helse- og omsorgsdepartementet.
Direktoratet har overtatt samtlige forvaltningsoppgaver etter bioteknologiloven. Direktoratet kan i slike saker innhente uttalelser fra Bioteknologinemnda.
Sosial- og helsedirektoratet er også delegert myndighet til å forvalte genteknologiloven, men bare de delene som omhandler innesluttet bruk av genmodifiserte organismer, og som direktoratet også er tilsynsmyndighet for.
Direktoratet forvalter også lov om biobanker i forhold til overføring av biobankmateriale til utlandet (§ 10), endret, utvidet, eller ny bruk (§ 13) og andres tilgang til materiale i en biobank (§ 15).
Direktoratet godkjenner videre prosjekter som omhandler bruk av fostervev med mer etter transplantasjonsloven.
Direktoratet behandler også søknader om fritak fra taushetsplikt i forskningsøyemed etter forvaltningsloven § 13 d og helsepersonelloven § 29 første ledd. Dispensasjon fra taushetsplikten er ikke nødvendig dersom pasientens samtykke innhentes eller dersom opplysningene gis i anonymisert form. 108 Anonymiseringen må da være utført på en slik måte at opplysninger ikke kan føres tilbake til enkeltpersoner. Dersom søknaden ikke er kurant, skal søknaden forelegges Rådet for taushetsplikt og forskning, jf. punkt 18.2.7 nedenfor. I praksis brukes nok Rådet mindre enn forutsatt.
18.2.3 Statens legemiddelverk
Statens legemiddelverk 109 (Legemiddelverket) er forvaltningsorganet på legemiddelområdet. Legemiddelverket skal ivareta forbrukernes og helsevesenets behov for effektive og sikre legemidler, samt bidra til riktig og rasjonell legemiddelbruk. Etaten fører tilsyn med produksjon, utprøving og omsetning av legemidler. 110 Statens legemiddelverk er et uavhengig forvaltningsorgan, administrativt underlagt Helse- og omsorgsdepartementet.
Forhåndskontroll
Alle kliniske utprøvinger av legemidler på mennesker , både på pasienter og friske forsøkspersoner skal meldes til Statens legemiddelverk, jf. forskrift om klinisk utprøving av legemidler til mennesker, fastsatt av Sosial- og helsedepartementet 24. september 2003 med hjemmel i legemiddelloven. Forskriften innarbeider EU direktiv (2001/20/EC) i norsk rett. Hensikten med direktivet er å harmonisere gjennomføringen av kliniske studier i hele EØS-området.
Legemiddelverkets vurderinger skjer på grunnlag av meldingen, som blant annet skal omfatte en vurdering av preparatets kvalitet, sikkerhet og antatte effekt, den vitenskapelig standard, forsøksmetoder, klinisk relevans og kvalitetssikring, jf. forskriftens kapittel 2 og 3 (§ 3-1). Er Statens legemiddelverks vurdering negativ, kan sponsor i henhold til § 3-1 annet ledd ”endre innholdet i søknaden én gang for å ta hensyn til innvendingen(e). Endres ikke søknaden i overensstemmelse med innvendingen(e), anses den for å være avvist, og det kliniske forsøk kan ikke starte.”
I forskriftens §5-2 står det også at kliniske utprøvinger skal skje i samsvar med Helsinki-deklarasjonen og dens senere revisjoner, samt at utprøvingen skal utføres i tråd med europeiske retningslinjer for Good Clinical Practice (GCP) 111 . Good Clinical Practice er standarder som skal sikre at de dataene som utprøvingen resuterer i, er pålitelige. De tre regelverkene er stort sett, men ikke helt, sammenfallende.
Vurdering av kliniske utprøvinger skal være avsluttet innen 60 dager fra komplett søknad ble innsendt (jf §3-2 første ledd). Dersom utprøvingen involverer genterapi, somatisk celleterapi og alle legemidler som inneholder genetisk modifiserte organismer heter det at ”Denne fristen kan forlenges én gang med 30 dager. Behandlingstiden på 90 dager kan forlenges med ytterligere 90 dager, dersom en gruppe eller en komité må konsulteres.”
Dersom Statens legemiddelverk ikke har avgitt sin vurdering innen fristen på 60 dager, kan utprøvingen starte, forutsatt at en regional komité for forskningsetikk (REK) har gitt en avsluttet positiv vurdering, jf. § 3-3. I følge § 1-5 skal alle kliniske utprøvinger ”vurderes av REK før de inngangsettes… Ingen studier kan settes i gang før det foreligger en positiv vurdering av meldingen fra REK”. Se om REK nedenfor.
Tilsyn
I følge forskriftens § 6-1 fører Legemiddelverket tilsyn med at bestemmelsene i forskriften overholdes. Enhver som er eller har vært involvert i gjennomføringen av utprøvinger av legemidler kan inspiseres, jf. forskriftens § 6-2. Inspeksjonen kan utføres på alle stadier av utprøvingen og også etter avsluttet utprøving. Inspeksjonsobjektene plukkes ut av en tverrfaglig gruppe ved Legemiddelverket. De aller fleste inspeksjonene er rutineinspeksjoner og er ”tilfeldig” plukket ut, dog etter ønske om å spre seg i forhold til legemiddelfirmaer, terapeutiske områder, utprøvere samt produktets utviklingsfase. I Norge har man valgt å føre tilsyn med sponsorer 112 (eller contract research organisations som har fått delegert sponsorforpliktelser) og utprøvere/utprøvingssteder. I andre land har man også ført tilsyn med andre aktører som f. eks. etikkomitéer.
I medhold av forskriftens § 6-3 kan en igangsatt utprøving ”på ethvert tidspunkt stanses av Statens legemiddelverk av hensyn til pasientsikkerhet, når utprøvingen ikke skjer i samsvar med gjeldende bestemmelser eller dersom Statens legemiddelverk finner det nødvendig av andre særlige grunner.”
I forskriftens kapittel 5 pålegges utprøver en rekke meldeplikter til Legemiddelverket, bl.a. ved uønskede medisinske hendelser og ved endringer av utprøvingen eller legemidlet.
Etterhåndskontroll
Innen 90 dager etter at studien er avsluttet, plikter sponsor iht. forskriftens § 5-9 tredje og fjerde ledd, ”å melde fra til Statens legemiddelverk og REK om at studien er avsluttet. Senest ett år etter at utprøvingen er avsluttet skal sponsor sende inn sluttrapport til Statens legemiddelverk.” En ny undersøkelse viser imidlertid at den pålagte sluttrapporteringen til Legemiddelverket svikter. Sju år senere var sluttrapport innsendt slik forskriftene krever, for under en fjerdedel av 208 legemiddelutprøvinger som ble meldt i 1996. 113
18.2.4 Datatilsynet
Datatilsynet 114 har siden opprettelsen i 1980 hatt som oppgave å beskytte den enkelte mot at personverninteressene krenkes gjennom måten personopplysninger behandles på. Personopplysninger skal behandles i samsvar med grunnleggende personvernhensyn, som behovet for vern av personlig integritet og privatlivets fred.
Det juridiske grunnlaget for Datatilsynets virksomhet er regulert i personopplysningsloven og helseregisterloven.
Datatilsynet er et uavhengig forvaltningsorgan, administrativt underordnet Moderniseringsdepartementet. Datatilsynets uavhengighet innebærer at departementet ikke kan gi instruks om, eller omgjøre Datatilsynets utøving av myndighet etter personopplysnings- eller helseregisterloven. Som klageinstans i forhold til Datatilsynets vedtak er det opprettet en personvernnemnd.
Personopplysningsloven legger opp til at det i første rekke er opp til hver enkelt å ivareta sitt eget personvern. I tillegg har de som behandler personopplysninger fått et større ansvar enn de hadde etter den gamle personregisterloven. Som en følge av dette er Datatilsynets oppgaver i stor grad gått fra forhåndsgodkjenning til etterkontroll.
Forhåndskontroll
Datatilsynet fører en offentlig fortegnelse over alle behandlinger av personopplysninger som er meldt inn. Disse blir kun registrert og ikke kontrollert (gjennomgått). De danner dog grunnlag for senere stikkprøver. Videre behandler Datatilsynet søknader om konsesjon, der dette kreves etter loven.
Datatilsynet skal behandle søknader om å opprette registre med sensitiv helseinformasjon så lenge registeret ikke er regulert i forskrift. Det er også lagt til grunn at Datatilsynet har mandat til å gi konsesjoner og administrativt avgjøre opprettelsen av store, sensitive helseregistre. Det betyr at alle private og de fleste offentlige prosjekter skal vurderes av Datatilsynet.
Noen forskningsprosjekter er pålagt meldeplikt i stedet for konsesjonsplikt gjennom forskriften til personopplysningsloven § 7-25.
Høsten 2004 sendte Datatilsynet ut på høring en forskrift som innebærer at Datatilsynet i tiden fremover, ser for seg mer tilsyn og mindre forhåndskontroll. Datatilsynet ser for seg at de bare bør føre forhåndskontroll med større forskningsprosjekter. Dette er i tråd med utvalgets anbefalinger, se kapittel 32.
Tilsyn
Gjennom aktivt tilsyn og saksbehandling kontrollerer Datatilsynet at lover og forskrifter for behandling av personopplysninger blir fulgt, og at feil og mangler blir rettet.
18.2.5 Statens helsetilsyn
Statens helsetilsyn 115 (Helsetilsynet) er et uavhengig forvaltningsorgan administrativt underlagt Helse- og omsorgsdepartementet. De fleste oppgavene er lagt direkte til Helsetilsynet i lov. Fylkeslegene opptrer som ”Helsetilsynet i fylkene”, og hører som sådan direkte under Helsetilsynet.
Statens helsetilsyn har røtter tilbake til 1809, da Det norske sundhedskollegiet ble opprettet. Det tidligere Helsedirektoratet ble omorganisert og endret til Statens helsetilsyn i 1992, med fokus på tilsyn og rettssikkerhet i helsetjenesten. Ved omorganiseringen 1. januar 2002, ble tilsynsprofilen styrket, mens statlige handlingsplaner og mange forvaltningsoppgaver m.m. ble overført til det nye Sosial- og helsedirektoratet.
Statens helsetilsyn har det overordnede faglige tilsyn med helsetjenesten. Dette fremgår av lov om statlig tilsyn med helsetjenesten (helsetilsynsloven) § 1.
Statens helsetilsyn og Helsetilsynet i fylkene fører tilsyn med all helsetjeneste og alt helsepersonell, jf. helsepersonelloven § 3. Det løpende (direkte) tilsynet med helsetjenesten og helsepersonell utøves av Helsetilsynet i fylkene som ledes av fylkeslegen. Dette fremgår av helsetilsynsloven §§ 2 og 3.
I følge helsetilsynsloven § 3 andre ledd skal Helsetilsynet i fylket ”påse at alle som yter helsetjenester har etablert internkontrollsystem og fører kontroll med sin egen virksomhet på en slik måte at det kan forebygge svikt i helsetjenesten.”
Forskningsaktiviteter som omfatter helsefremmende tiltak, diagnose og behandling, habilitering og rehabilitering, pleie og omsorg og opplæring av pasienter og pårørende, eller som utføres av helsepersonell i kraft av deres autorisasjon, omfattes av det tilsyn som Statens helsetilsyn og Helsetilsynet i fylkene fører. Det vil altså si terapeutisk forskning. Hvorvidt tilsyn med ikke-terapeutisk forsk-ning omfattes av Helsetilsynets virksomhet er noe uklart. Det ser ut til å være en utbredt oppfatning at personer autorisert som helsepersonell anses som helsepersonell nærmest uansett hvilke oppgaver utfører, og særlig dersom dette foregår på et sykehus eller annet sted hvor det også regelmessig ytes helsetjenester. En slik utvidende tolkning har mye for seg. Blant annet kan det for pasienter og andre forskningsdeltakere være vanskelig å skille mellom lege (helsepersonell) og forsker (ikke-helsepersonell som ikke yter helsetjenester). Selv leger og annet helsepersonell og tilsynsorganer vil kunne ha problemer med det.
Etter helsetilsynsloven § 5 har Statens helsetilsyn adgang til å gi pålegg til helsetjenesten ”hvis virksomhet innen helsetjenesten drives på en måte som kan ha skadelige følger for pasienter eller andre eller på annen måte er uheldig eller uforsvarlig.”
Statens helsetilsyn eller Helsetilsynet i fylkene har hittil i liten grad gjennomført planlagte tilsyn med forskning innen helsetjenesten. Forskningsaktiviteter og -prosjekter har imidlertid vært tema ved flere hendelsesbaserte tilsyn. Det vil si tilsyn på bakgrunn av hendelser.
18.2.6 Domstolskontroll
Domstolene skiller seg fra de nevnte forvaltningsorganer ved at de hører inn under den dømmende makt og ikke er en del av den utøvende makt. Domstolene har som en av sine viktigste funksjoner å avgjøre rettstvister borgerne mellom og mellom borgerne og offentlige myndigheter. Domstolene anvender den retten som er skapt av lovgiver (Stortinget). De avgjør de enkelttilfellene de får seg forelagt på grunnlag av gjeldende rett. Lov og andre rettskilder gir ikke alltid noe sikkert svar på de rettsspørsmål som oppstår, og da må domstolene selv finne frem til løsningen og utøve et såkalt rettslig skjønn. Ved avgjørelsen av et rettsspørsmål det er tvil om vil hensynet til hva som er rettferdig, (etisk) forsvarlig og hensiktsmessig ofte spille en vesentlig rolle.
Borgere som mener at de har lidd urett, kan anlegge sak for domstolene. Det betyr for eksempel at skadelidte forsøkspersoner kan anlegge erstatningssak mot ansvarlig forsker/forskningsinstitusjon. Dermed fører domstolene såkalt domstolskontroll med forskere.
Dersom for eksempel forskere ikke er enig i forvaltningsorganers rettsanvendelse, kan forskere bringe saken inn for domstolene. Dermed fører domstolene også kontroll med forvaltningen.
I Norge har imidlertid ikke domstolene spilt noen sentral rolle som kontrollorganer for medisinsk forskning, i motsetning til noen andre land, særlig USA. Norske domstoler har heller ikke vært sentrale ved utviklingen av gjeldende rett på dette området, slik de for eksempel har vært det innenfor erstatningsretten. Grunnen til at svært få saker vedrørende medisinsk forskning blir brakt inn for domstolene, kan være at det sjelden oppstår skader ved forskning, eller at det ellers ikke er behov for å få overprøvd omtalte forvaltningsorganers avgjørelser. Det kan igjen tolkes som at forskning i Norge i hovedsak utøves forsvarlig og at forskerne innfinner seg med forvaltningsorganenes myndighetsutøvelse. Det siste kan være tilfelle ved at de fleste forvaltningsorganer ofte legger opp til en dialog med forskeren, jf. veiledningsplikten, slik at forsker og forvaltningsorgan finner frem til en løsning. Andre årsaker kan være at så vel forskere som forsøkspersoner kvier seg for å bringe en sak inn for domstolene.
18.2.7 Rådet for taushetsplikt og forskning
Rådet for taushetsplikt og forskning er et uavhengig forvaltningsorgan administrativt underlagt Justisdepartementet. Rådets virksomhet er hjemlet i forskrift til forvaltningsloven § 13 d.
I henhold til forvaltningsloven § 13 d (og helsepersonelloven § 29) kan vedkommende fagdepartementet bestemme at opplysninger kan eller skal utleveres til bruk i forskning, og at slik utlevering kan skje uten hinder av taushetsplikt.
Bestemmelsene gjelder dispensasjon fra henholdsvis yrkesmessig og forvaltningsmessig taushetsplikt. Søknad om dispensasjon fra yrkesmessig taushetsplikt vil som regel gjelde innsyn i journalopplysninger. Søknad vedrørende den forvaltningsmessige taushetsplikt vil som regel gjelde innsyn i helseregistre (Medisinsk fødselsregister, Kreftregisteret, Dødsårsaksregisteret m.v.). Helse- og omsorgsdepartementet har delegert sin myndighet på helseområdet til Sosial- og helsedirektoratet.
Før direktoratet treffer vedtak om å gi eller å avslå en begjæring om å få opplysninger undergitt taushetsplikt til bruk for forskning, skal saken forelegges Rådet for taushetsplikt og forskning.
Det er ikke nødvendig å forelegge saken for rådet dersom saken er kurant. Hva som er kurant, vil i stor utstrekning være opp til den som dispenserer (her: Sosial- og helsedirektoratet) å avgjøre. I forskriftene er det bestemt at der hvor forskeren skal ta direkte kontakt med de personene som de taushetsbelagte opplysningene angår, så skal saken i alle tilfelle forelegges rådet. Det er altså forvaltningsorganet selv, og ikke forskeren, som skal kontakte rådet før dispensasjonssøknaden avgjøres.
Rådet har fire medlemmer og en sekretær, som alle er oppnevnt av Justisdepartementet.
Rådet skal i henhold til forskrift til forvaltningslovens § 13 d avgi uttalelse før forvaltningen fatter vedtak om å gi dispensasjon fra taushetsplikten. Dette er formelt sett et samtykke, noe som innebærer at det ikke er rådet som fatter den endelige beslutningen om å gi eller ikke gi dispensasjon fra taushetsplikten. 116 Bare Kongen i statsråd kan frita for taushetsplikt i strid med rådets uttalelse.
Loven er særdeles åpen med hensyn til kriteriene for når dispensasjon kan gis. Utfallet beror da også på en skjønnsbasert helhetsvurdering, hvor det på den ene side må tas hensyn til forskning, og på den andre hensynet til den som taushetsplikten i det konkrete tilfelle tar sikte på å verne.
Det følger av forvaltningsloven § 13 e at forskeren har taushetsplikt på samme måte som tjenestemenn i forvaltningen når forskeren mottar opplysningene.
Saksbehandlingen hos rådet er skriftlig og tar omtrent én måned. I dag er det relativt sjelden at rådet mottar saker fra direktoratet. Totalt (dvs. innbefattet alle områder, ikke bare helse) behandler rådet ca. 20 saker årlig. Justisdepartementet har i forbindelse med en opprydding i forskriftene til forvaltningsloven sendt ut på høring et forslag som innebærer at rådet fortsetter sin virksomhet i nåværende form. 117 I utkastets § 9 heter det at:
Før det blir truffet vedtak om å gi opplysninger undergitt taushetsplikt til bruk for forskning eller å avslå en begjæring om å få slike opplysninger, skal saken forelegges for Rådet for taushetsplikt og forskning. Bare Kongen kan frita for taushetsplikt i strid med Rådets uttalelse.
Dersom departementet eller et annet organ med myndighet som nevnt i § 8 finner det klart at søknaden bør innvilges eller avslås, behøver saken ikke forelegges for Rådet. Ved avgjørelsen av om en sak ikke skal forelegges for Rådet, skal det særlig legges vekt på om de opplysninger det søkes om tilgang til, må anses som følsomme. Ellers bør det legges vekt på om den materialet skal stilles til rådighet for, har betryggende faglig kompetanse eller er undergitt forsvarlig faglig veiledning.
Saken skal i alle tilfelle forelegges Rådet dersom forskeren skal ta direkte kontakt med de personene opplysningene angår.
18.2.8 Bioteknologinemnda
Bioteknologinemnda 118 er et frittstående, regjeringsoppnevnt organ for forvaltningen og ble første gang oppnevnt i 1991. Nemnda er hjemlet i lov om medisinsk bruk av bioteknologi og lov om fremstilling og bruk av genmodifiserte organismer. I mandatet for 2000 - 2004 heter det:
Bioteknologinemnda er et rådgivende og frittstående organ for forvaltningen som særlig skal vurdere og drøfte prinsipielle eller generelle spørsmål knyttet til bioteknologi og genteknologi, herunder samfunnsmessige og etiske spørsmål.
Bioteknologinemnda skal legge stor vekt på informasjons- og debattskapende aktiviteter ved å bidra til informasjon til publikum og bidra til kommunikasjon mellom offentlige myndigheter, fagfolk og interesseorganisasjoner.
Bioteknologinemnda skal på begjæring eller av eget tiltak gi uttalelser i saker etter lov om humanmedisinsk bruk av bioteknologi m.m. og lov om fremstilling og bruk av genmodifiserte organismer, herunder forslag til endringer i lov, forskrifter mv. som har betydning for bioteknologi.
Bioteknologinemnda skal gi uttalelser til norske myndigheter som angår Norges holdning til spørsmål vedrørende bioteknologi i internasjonale organer.
Bioteknologinemnda skal videre belyse spørsmål som er særlig viktig i et Nord/Sør-perspektiv.
Bioteknologinemndas uttalelser er offentlige med mindre annet følger av lovbestemt taushetsplikt.
Humanmedisinsk bruk av bioteknologi skal forelegges bioteknologinemnda før søknad om godkjennelse sendes Sosial- og helsedirektoratet, jf. bioteknologiloven §§ 2-19, 4-2, 5-3 og 6-3,1.
Bioteknologinemnda har 24 medlemmer og observatører fra seks departementer.
Nemnda gir råd til forvaltningen (det vil si til Helse- og omsorgsdepartementet eller Sosial- og helsedirektoratet). Forskere vil dermed ikke ha direkte kontakt med nemnda.
18.2.9 Personvernnemnda
Personvernnemnda er opprettet med hjemmel i personopplysningsloven § 43. Personvernnemnda skal behandle klager på vedtak som Datatilsynet fatter i medhold av personopplysningsloven og enkelte andre lover.
Personvernnemnda er et uavhengig forvaltningsorgan administrativt underlagt Moderniseringsdepartementet. Regler i forvaltningsloven og offentlighetsloven kommer også til anvendelse på nemndas arbeid. Departementet har utarbeidet instruks for nemnda, men dette innebærer ikke noen form for instruksjonsmyndighet i enkeltsaker. Departementet kan heller ikke gi generelle instrukser om lovtolkning eller skjønnsutøvelse.
Personvernnemnda har sju medlemmer og skal årlig orientere Kongen om behandling av klagesakene. 119
18.3 Myndighetsnære organer
18.3.1 De regionale komiteer for medisinsk forskningsetikk (REK) og Den nasjonale forskningsetiske komité for medisin (NEM)
Generelt
De regionale komiteer for medisinsk forskningsetikks (REK) oppgave er å veilede og gi råd om forskningsetiske spørsmål, og å arbeide for å gjøre forskningsetiske prinsipper kjent. 120
Komiteene (totalt fem) har i praksis en meget sentral kontrollfunksjon ved medisinsk og helsefaglig forskningsvirksomhet, ved at stort sett alle medisinske forskningsprosjekter som involverer mennesker og humant biologisk materiale, forskning på lik og registerdata skal forelegges komiteene.
Komiteene er imidlertid formelt sett ikke forvaltningsorganer. Komiteenes virksomhet innebærer imidlertid reell myndighetsutøvelse, som i enkelte tilfeller er hjemlet i lov, slik at det er nærliggende å karakterisere komiteene som ”myndighetsnære organer”, se nedenfor.
Bakgrunn
Bakgrunnen for opprettelsen av de regionale komiteene for medisinsk forskningsetikk (forkortet REK) i Norge, er en bestemmelse i Helsinki-deklarasjonen. Gjeldende artikkel 13 lyder:
Opplegget for og utførelsen av ethvert eksperiment som omfatter mennesker må være klart formulert i en forsøksprotokoll. Denne protokollen skal legges frem for en spesielt oppnevnt etisk komité for vurdering, uttalelse, veiledning og eventuelt godkjennelse. Komiteen skal være uavhengig av forsker og sponsor og andre som kan ha utilbørlig innflytelse. Denne uavhengige komiteen skal være i samsvar med lovgivningen og rettsreglene i det landet hvor forskningen utføres. Komiteen har rett til å overvåke pågående forsøk. Forskeren har plikt til å gi komiteen nødvendige opplysninger ved oppfølging, særlig gjelder dette alvorlige hendelser. Forskeren skal også legge frem for komiteen til vurdering opplysninger om finansiering, sponsorer, institusjonstilhørighet, andre potensielle interessekonflikter og incitamenter for forsøkspersoner.
Norge var forholdsvis sent ute med å etablere slike komiteer; dette skjedde først i 1985. Før det hadde lokale komiteer ved enkelte sykehus anvendt Helsinki-deklarasjonens regler på ad hoc basis.
Hjemmelsgrunnlag - organisering
Komiteenes mandat er fastsatt av Utdannings- og forskningsdepartementet. Det er opprettet fem regionale komiteer, en i hver helseregion; REK Sør, REK Vest, REK Midt-Norge, REK Nord og REK Øst. Komiteenes administrasjon er knyttet til de fire medisinske fakulteter ved universitetene i Oslo, Bergen, Trondheim og Tromsø. Bevilgningene skjer over fakultetenes budsjett, og komiteene er administrativt tilknyttet fakultetene. Den nasjonale forskningsetiske komité for medisin (NEM) er rådgivende og koordinerende instans for de regionale komiteer for medisinsk forskningsetikk, og skal ta initiativ til å følge den forskningsetiske debatten.
Departementet oppnevner medlemmene, og fastsetter hvem som skal være leder og nestleder. Før oppnevnelsen innhentes det uttalelse fra flere, blant annet de medisinske fakultetene. Oppnevnelsen skjer for en periode av fire år, og ingen kan oppnevnes for mer enn to perioder sammenhengende. Komiteene har åtte medlemmer hver og består av:
En medisinsk fagkyndig fra det medisinske fakultet i regionen
en medisinsk fagkyndig fra den offentlige helsemyndighet i regionen
en psykologisk fagkyndig fra det psykologiske institutt eller fakultet i regionen
en offentlig godkjent sykepleier
et medlem fra sykehuseierne i regionen
et medlem med fagkyndighet i etikk
en jurist, og
en lekrepresentant.
Selv om komiteene er oppnevnt av departementet, er de ikke organisert som forvaltningsorganer. Komiteene har status som frie og uavhengige organer. Det vil si at ingen andre kan instruere komiteene i deres virksomhet. Komiteenes øvrige status og rolle er imidlertid uklar.
Komiteenes ”råd” er ikke rettslig bindende. Det er således i prinsippet opp til forskeren om han/hun vil følge komiteens råd. Men i praksis gjøres dette så godt som alltid, da det er en del indirekte konsekvenser av å unnlate å følge rådet. For eksempel forutsetter i praksis Datatilsynet, Statens legemiddelverk og Helse- og omsorgsdepartementet at komiteene har tilrådd prosjektet. I tillegg vil man neppe få publisert resultatet i et anerkjent tidsskrift i henhold til Vancouver-konvensjonen, 121 som sentrale medisinske tidsskrifter verden over har sluttet seg til. Dermed fremstår komiteens tilrådning reelt sett for alle praktiske formål, som en forhåndstillatelse – det vil si myndighetsutøvelse.
Komiteenes reelle myndighet er uansett stor, og det er nærliggende å karakterisere dem som ”myndighetsnære organer”.
Komiteene treffer i følge komiteenes mandat ikke ”enkeltvedtak”. I forvaltningsloven er enkeltvedtak definert som ”en avgjørelse som treffes under utøving av offentlig myndighet og som generelt eller konkret er bestemmende for rettigheter eller plikter til private personer (enkeltpersoner eller andre private rettssubjekter)”(§ 2). Fordi mandatet bestemmer at komiteene ikke treffer enkeltvedtak, gjelder ikke forvaltningslovens regler om klage og begrunnelse av vedtak for komiteenes virksomhet.
Virksomhetsområde
I komiteenes mandat er komiteens virksomhetsområde angitt meget vidt. I mandatet punkt 3 heter det:
Komiteene skal forelegges samtlige biomedisinske forskningsprosjekter hvor det inngår forsøk på mennesker og som ikke er av en slik art at det regnes som en del av vanlig etablert behandlingsprosedyre. Det gjelder både terapeutisk og ikke-terapeutisk forskning på pasienter og friske forsøkspersoner. Med biomedisinske forskningsprosjekter hvor det inngår forsøk på mennesker forstås også forskning på identifiserbart eller anonymt humant materiale og identifiserbare eller anonyme data. Bestemmelsene her gjelder tilsvarende for forsøk på lik og for forsøk på eller med fostre, samt fostervev.
I kommentarene til mandatet heter det at ”biomedisinsk forskning forstås i denne sammenheng noe videre enn hva tradisjonen har vært. Det kan omfatte forskning som anvender psykologiske, samfunnsvitenskapelige og bioteknologisk metodikk. Kriteriene for at slike forskningsprosjekter skal være fremleggelsespliktige, er at de omhandler menneskers fysiske eller mentale helse og anvender terapeutiske eller ikke-terapeutiske metoder på personer.”
Selv om det i komiteenes mandat er fastslått at alle medisinske forsøk skal forelegges komiteene, er det allikevel ingen rettslig forpliktelse til å gjøre dette. Det vil således bare være snakk om et etisk imperativ. Det er imidlertid som nevnt etter hvert etablert betydelige reelle og formelle sanksjoner ved unnlatelse av komitébehandling. Rent faktisk vil derfor de aller fleste medisinske forskningsprosjekter bli forelagt REK.
I tillegg har det i de siste årene vært en tendens til å sette krav om komitébehandling i enkelte lover og forskrifter, jf. for eksempel biobankloven § 4 og forskrift om klinisk utprøving av legemidler av 2003 § 1-5. For eksempel fastsetter legemiddelforskriften i § 1-5 annet ledd hvilke vurderinger komiteene skal gjøre. I tredje ledd fastsettes enkelte saksbehandlingsrutiner. Man får dermed en slags indirekte lovhjemling av komitésystemet, uten at det er fastsatt nærmere hvordan komiteene skal forholde seg til øvrige reguleringer på området som de tidligere har forholdt seg til, og som de vil forholde seg til i andre saker.
Myndighetsområde
I mandatets punkt 2 heter det at ”Komiteene gir råd og veiledning etter en alminnelig forskningsetisk vurdering, hvor det også tas hensyn til forskningsetiske retningslinjer vedtatt av nasjonale og internasjonale organer (f.eks. Helsinki-deklarasjonen).” I kommentarer til mandatet heter det at eksempler på vanlige forskningsetiske prinsipper er kravet om frivillig informert samtykke fra forsøkspersoner, at skillet mellom terapeutisk og ikke-terapeutisk forskning er relevant, og at mulig risiko og ubehag for forsøkspersoner skal avveies mot den medisinske betydning forsøksprosjektet har for forsøkspersonen selv og/eller andre.
I standard prosedyrer for saksbehandling i De regionale komiteer for medisinsk forksningsetikk 122 (Standard Operating Procedures (SOP)) heter det at saksbehandlingen i komiteene er basert på
..mandatet, Helsinkideklarasjonen, Vancouver-konvensjonen og veiledningen til skjema.
Mandatet understreker at komiteene skal foreta en alminnelig forskningsetisk vurdering, hvor det også tas hensyn til forskningsetiske retningslinjer av nasjonale og internasjonale organer. Blant utenlandske organer henvises det særlig til internasjonale retningslinjer utgitt av Council of International Organizations of Medical Sciences som både har utgitt en veiledning om forskning på mennesker generelt (International Guidelines for Biomedical Research Involving Human Subjects) og om epidemiologiske studier spesielt (International Guidelines for Ethical Review of Epidemiological Studies).
Ved kliniske utprøvinger baserer vurderingene seg også på Forskrift om klinisk utprøving av legemidler til mennesker og på retningslinjer for god klinisk prøvningspraksis – Guideline for Good Clinical Practice - som har til hensikt å skape en enhetlig vitenskapelig og etisk standard for klinisk forskning. Disse retningslinjene har Norge sluttet seg til gjennom EØS-samarbeidet på grunnlag av EU-direktiv 2001/20/EC om iverksettelsen av god klinisk prøvningspraksis. REK har tatt forbehold om noen administrative krav i kapittel 3 i International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) sine retningslinjer for god klinisk utprøvingspraksis (GCP), 1996.
Det fragmenterte og omfattende, dels skrevne og dels uskrevne, dels nasjonale og dels internasjonale, regelverket komiteene bygger sine avgjørelser på, gjør det vanskelig for forskere å forutsi sin rettsstilling. Fra et rettsikkerhetsperspektiv er det problematisk.
I 2001 ble komiteenes mandat endret slik at forvaltningslovens regler om habilitet og alminnelige regler om saksbehandlingen og om partsinnsyn ble gjort gjeldende. Det ble presisert at komiteene ikke fatter enkeltvedtak etter forvaltningsloven og at lovens regler om begrunnelse og klage ikke skal gjelde. Offentlighetsloven gjelder i sin helhet.
Det er altså ikke etablert en formell klageordning. Klageadgang anses normalt som en helt fundamental rettsikkerhetsgaranti. REK har imidlertid, etter anmodning fra Den nasjonale forskningsetiske komité for medisin (NEM), pålagt seg selv å gi mulighet for fornyet behandling i en annen regional komité når det foreligger en frarådning.
18.3.2 Uredelighetsutvalg
Fra 1994 til 2001 eksisterte det et utvalg opprettet av Norsk forskningsråd, kalt Nasjonalt utvalg for vurdering av uredelighet i helsefaglig forskning. Utdannings- og forskningsdepartementet anbefaler i et lovutkast at dette gjenopprettes, dog i en noe endret form.
18.4 Frie aktører
18.4.1 Personvernombud for forskning - særlig om Norsk samfunnsvitenska-pelig datatjeneste (NSD)
Generelt om Personvernombud
I følge § 7-12 i forskrift til personopplysningsloven kan Datatilsynet
samtykke i at det gjøres unntak fra meldeplikt etter personopplysningsloven § 31 første ledd, dersom den behandlingsansvarlige utpeker et uavhengig personvernombud som har i oppgave å sikre at den behandlingsansvarlige følger personopplysningsloven med forskrift. Personvernombudet skal også føre en oversikt over opplysningene som nevnt i personopplysningsloven § 32.
Se om dette i kapittel 14 om personvern- og personopplysningsrett ovenfor.
Hvem som helst kan dermed i prinsippet utnevnes som personvernombud. For eksempel har Ullevål sykehus oppnevnt eget personvernombud.
Særlig om Norsk samfunnsvitenskapelig datatjeneste AS
Norsk samfunnsvitenskapelig datatjeneste AS (NSD) 123 er et privat foretak som tilbyr enkelte tjenester til forskere og forskningsinstitusjoner mot betaling. Det er altså frivillig hvorvidt den enkelte forsker/forskningsinstitusjon vil benytte seg av NSD sine tjenester.
En av de tjenester NSD tilbyr, er å være personvernombud for forskere. Svært mange forskere/forskningsinstitusjoner benytter seg av denne tjenesten, noe som er med på å bygge opp under et inntrykk av at NSD er et forvaltningsorgan eller er i hvert fall et organ som driver med en form for myndighetsutøvelse. Det er imidlertid ikke riktig. Personvernombudet driver ikke offentlig myndighetsutøvelse. NSD skal hjelpe forskeren å ivareta personvernet til personer som deltar i forskningsprosjekter.
NSD er i dag personvernombud for alle universitetene, de statlige høyskolene, de vitenskapelige og private høyskolene, en rekke helseforetak og andre forskningsinstitusjoner. Forskere og studenter som er tilknyttet disse institusjonene og som skal gjennomføre prosjekt som medfører behandling av personopplysninger, har meldeplikt til Personvernombudet for forskning i stedet for til Datatilsynet.
I følge NSD er personvernombudets hovedoppgaver er å:
Vurdere forsknings- og studentprosjekt i forhold til bestemmelsene i personopplysningsloven og helseregisterloven med tilhørende forskrifter.
Gi informasjon og veiledning til institusjonene og til den enkelte forsker og student om forskning og personvern.
Bistå den registrerte med å ivareta sine rettigheter.
Føre en systematisk og offentlig oversikt over alle behandlinger
NSD vurderer innkomne meldeskjemaer i henhold til gjeldende regelverk og avtaler. Ved uklarheter tar ombudet direkte kontakt med prosjektleder. Videre saksgang avhenger av om prosjektet er underlagt meldeplikt eller konsesjonsplikt i henhold til personopplysningsloven.
Meldeplikt er hovedregelen i personopplysningsloven. Dersom prosjektet blir vurdert som meldepliktig, sender NSD (personvernombudet) skriftlig tilbakemelding på undersøkelsesopplegget til prosjektleder. I tilbakemeldingen vil det gå frem om prosjektet oppfyller kravene i personopplysningsloven og eventuelt helseregisterloven og om datainnsamlingen kan starte.
Dersom forskningsprosjektet omfatter behandling av sensitive opplysninger (for eksempel helseopplysninger), vil prosjektet som hovedregelen være underlagt konsesjonsplikt. NSD som personvernombud vurderer undersøkelsesopplegget og lager en innstilling som sendes til Datatilsynet sammen med all relevant dokumentasjon. Prosjektleder får tilsendt kopi av innstillingen. Datatilsynets vedtak sendes direkte til prosjektleder. Kopi av vedtaket sendes til NSD. Prosjektleder kan ikke begynne datainnsamlingen før konsesjon fra Datatilsynet foreligger. Ordningene med NSDs deltakelse i Datatilsynets saksbehandling er ikke nærmere regulert i lov eller forskrift.
18.4.2 Forskningsutvalg og andre frie aktører
I figur 18.1 er institusjon/arbeidsgiver, for eksempel universitet, sykehus, privat foretak, industri og lignende tatt med. Dette fordi de ofte spiller en mer eller mindre sentral rolle ved kontroll og tilsyn med medisinsk og helsefaglig forskning. Forskere er som regel knyttet til en institusjon eller foretak som arbeidstaker.
Finaniseringerskilde, oppdragsgiver og lignende er også tatt med i figuren. Disse er gjerne forskjellig fra institusjon/arbeidsgiver.
Felles for dem er at de ofte spiller en mer eller mindre sentral rolle ved kontroll og tilsyn med medisinsk og helsefaglig forskning. For eksempel vil finansieringskilde stille krav til forskerens kompetanse, studiets design, gjennomføring osv. Slik kvalitetssikring, internkontroll eller lignende, vil kunne minske behovet for ekstern offentlig kontroll.
19 Reguleringen i Danmark og Sverige
19.1 Innledning
Andre lands reguleringer har liten direkte betydning for reguleringene i Norge. De kan imidlertid være av stor betydning som sammenligningsgrunnlag, spesielt gjelder dette våre naboland Sverige og Danmark.
For det første tilstreber vi en viss harmonisering/likhet i regelverket bl.a. pga. internasjonalt samarbeid. Dessuten forholder vi oss til det samme internasjonale regelverk, og det kan ha interesse å se hvordan våre naboland innretter seg i forhold til det. Historisk er det også mange likhetstrekk mellom reguleringen av medisinsk og helsefaglig forskning i Norge, Sverige og Danmark. Dette har sammenheng med relativt samme rettstradisjon og utstrakt samarbeid.
I det følgende gis først en kort sammenlignede vurdering av reguleringene i Norge, Sverige og Danmark. Dernest omtales Danmark og Sverige spesielt.
19.2 Oversikt over likhetstrekk mellom Norge, Sverige og Danmark
Forskning på mennesker. Danmark vedtok egen lov for medisinsk forskning i 1992, mens Sverige fikk en tilsvarende lov i 2004. Felles for disse lovene er at de først og fremst lovhjemler etikkomiteesystemet. I tillegg slås enkelte sentrale forskningsetiske prinsipper fast. I alle land i EØS-området er Direktiv 2001/20/EC om klinisk utprøving av legemidler innarbeidet i landets regelverk. Det er derfor felles regelverk i alle nordiske land på dette området.
Forskning på humant biologisk materiale. Alle landene erkjente nylig behovet for en regulering av innsamling, bruk, oppbevaring mv. av humant biologisk materiale (biobankvirksomhet) og satte i gang utredningsarbeid parallelt. Biobanklover ble vedtatt i Sverige i 2002 og i Norge i 2003. I Danmark valgte man i stedet for å vedta en egen lov for å biobanker å supplere allerede eksisterende lover (pasientrettighetsloven og personopplysningsloven), slik at biobankvirksomhet også må anses regulert i Danmark.
Alle landene har regulert embryoforskning, stamcelleforskning og kloning. Kloning er forbudt i alle land. Embryoforskning er tillatt i inntil 14 dager i Sverige og Danmark, men ikke i Norge. Norges bioteknologilov setter også andre grenser som en ikke har i de andre nordiske landene (gjelder genetisk testing, genterapi).
Forskning på helseopplysninger. Alle landene har implementert EU’s personverndirektiv, og har egne personopplysningslover som ligner norsk lovgivning.
Byråkrati. Både i Sverige og Danmark må forskere, som i Norge, forholde seg til flere andre ulike forvaltningsorganer, samt etikkomiteene. I både Sverige og Danmark er etikkomtessystemet lovhjemlet, og det er etablert en klageadgang.
Hovedtrekk. Likhetstrekkene er mange mellom de tre land. Det eksisterer likevel i det som i det store og hele kan karakteriseres som ”enkelte nyanseforskjeller”, som gjør at særlig Danmark betraktes som mer liberalt enn i Norge fra forskerhold.
19.3 Danmark
19.3.1 Lovregulering av medisinsk og helsefaglig forskning
Danmark vedtok i 1992 en lov – lov av 24. juni 1992 om et videnskabsetisk komitesystem og behandling av biomedicinske forskningsprojekter. Loven gjelder ethvert biomedisinsk forsøk på:
levendefødte menneskelige individer,
menneskelige kønsceller, der agtes anvendt til befrugtning, menneskelige befrugtede æg, fosteranlæg og fostre,
væv, celler og arvebestanddele fra mennesker, fostre og lignende og
afdøde, skal anmeldes til den regionale komité for det område, hvori den projektansvarlige har sit virke.
Stk. 2. Det samme gælder forskningsprojekter, hvori biomedicinsk forskning som nævnt i stk. 1, udgør en væsentlig bestanddel af det samlede projekt.
Stk. 3. Spørgeskemaundersøgelser og registerforskningsprojekter skal kun anmeldes til en regional komité, såfremt projektet indeholder et væsentligt element af biomedicinsk forskning eller udgør en del af et projekt, som indeholder et væsentligt element af biomedicinsk forskning, og såfremt projektet for den enkelte forsøgsperson indebærer sundhedsmæssige risici eller på anden måde kan være til belastning for den pågældende forsøgsperson.
Stk. 4. Den centrale komité kan fastsætte regler for den praktiske tilrettelæggelse af den videnskabsetiske bedømmelse af projekter, der udføres på flere forskningsinstitutioner (multicenterundersøgelser).
I tillegg har Danmark personopplysningslovgivning og pasientrettighetslovgivning som er sentrale på dette området.
19.3.2 Særlig om komitésystemet
Danmark har i likhet med Norge mange ulike offentlige organer som forhåndsvurderer forskningsprosjekter.
Et system med regionale forskningsetiske komiteer ble etablert i 1980 i Danmark. I tillegg ble det opprettet en sentralkomite, Den Centrale Videnskabsetiske Komite (CVK). Systemets primære oppgave er å sikre beskyttelse av personer som deltar i biomedisinske forskningsprosjekter, samtidig som det skal utbre kjennskap til etiske problemstillinger.
Systemet var frivillig og ulovfestet frem til loven ble vedtatt i 1992. Komiteene er i dag forvalningsorganer som treffer enkeltvedtak. De er administrativt tilknyttet kommunene og oppnevnt av dem. Loven var hovedsakelig en kodifisering av gjeldende praksis.
Det forskningsetiske komitésystemet i Danmark består i dag av CVK og sju regionale komiteer, alle med representasjon av lekfolk og forskere. Komiteenes størrelse kan variere, men de skal ha minst sju medlemmer, hvorav et mindretall på tre må være aktive innenfor medisinsk og helsefaglig forskning, mens et flertall skal være lekfolk. Komiteene utarbeider selv forslag til vedtekter, som så godkjennes av den sentrale komiteen. Det tas et gebyr per prosjekt til delvis dekning av utgiftene.
Til forskjell fra det norske systemet, har komiteene ikke bare en rådgivende funksjon, men gir tillatelse eller avslår anmeldte prosjekter. De har dessuten en viktig kontrollfunksjon, dvs. at de skal føre tilsyn med at prosjektet utføres i overensstemmelse med tillatelsen. Dette innebærer at komiteen kan følge prosjektets gang og blant annet kreve å få tilsendt den avsluttende rapporten eller publikasjonen. Prosjektleder skal underrette komiteen om mulige bivirkninger eller alvorlige hendelser som inntreffer i løpet av forsøket, og vesentlige endringer i forsøksprotokollen må godkjennes av komiteen. Det er satt tidsfrister i loven for behandling av prosjekter og endringer i prosjekter.
Den sentrale komiteen, CVK, oppnevnes av den danske forskningsministeren. CVKs hovedoppgaver er å koordinere arbeidet i de regionale komiteene, fastsette veiledende retningslinjer og uttale seg om spørsmål av prinsipiell karakter. De prinsipielle spørsmålene – som reises uavhengig av enkeltsaker – kan for eksempel gjelde nye forskningsområder. CVK skal videre følge forskningsutviklingen og drive informasjon om forskningsetiske problemstillinger. CVK skal også behandle enkeltsaker. Hvis det ikke oppnås enighet i en regional komité, bringes saken inn for den sentrale komiteen til avgjørelse. Endelig skal CVK være klageinstans for avgjørelser fattet i de regionale komiteene. Klageordningen fungerer slik at en prosjektleder som har fått avslag i en regional komité, eller en annen som er part i saken, kan bringe avgjørelsen inn for CVK innen 30 dager. CVKs avgjørelse kan ikke klages videre. Ifølge årsmeldingen for 2001 behandlet CVK 43 enkeltsaker dette året, og sju av dem var klagesaker.
CVK er pålagt å samarbeide med Det Etiske Råd om prinsipielle etiske spørsmål. Dette rådets oppgave er å rådgi den danske helseministeren, Folketinget og helsemyndighetene om etiske forhold, særlig i forbindelse med ny biomedisinsk teknologi.
19.4 Sverige
19.4.1 Lovregulering av medisinsk og helsefaglig forskning
I 2003 ble ”lag om etikprövning av forskning som avser människor” vedtatt i Sverige. Loven trådte i kraft 1. januar 2004.
Formålet med loven er ”att skydda den enskilda människan och respekten för människovärdet vid forskning” (§ 1)
Loven inneholder bestemmelser om etikkprøving av forskning som involverer mennesker og humant biologisk materiale. Den inneholder også bestemmelser om samtykke til slik forskning.
I lovens § 2 finnes følgende definisjoner:
”forskning: vetenskaplig forskning samt utvecklingsarbete på vetenskaplig grund,
forskningshuvudman: en statlig myndighet eller en fysisk eller juridisk person i vars verksamhet forskningen utförs, och
forskningsperson: en levande människa som forskningen avser.
Lovens saklige virksomhetsområdet angis i § 3 og § 4 til å være:
”känsliga personuppgifter enligt 13 § personuppgiftslagen (1998:204), eller
personuppgifter om lagöverträdelser som innefattar brott, domar i brottmål, straffprocessuella tvångsmedel eller administrativa frihetsberövanden enligt 21 § personuppgiftslagen, om forskningspersonen inte har lämnat sitt uttryckliga samtycke till behandlingen.
4 § Utöver vad som följer av 3 § skall lagen tillämpas på forskning som
innebär ett fysiskt ingrepp på en forskningsperson,
utförs enligt en metod som syftar till att påverka forskningspersonen fysiskt eller psykiskt,
avser studier på biologiskt material som har tagits från en levande människa och kan härledas till denna människa,
innebär ett fysiskt ingrepp på en avliden människa, eller
avser studier på biologiskt material som har tagits för medicinskt ändamål från en avliden människa och kan härledas till denna människa.”
I §§ 7-11 angis utgangspunkter for etikkprøvingen:
7 § Forskning får godkännas bara om den kan utföras med respekt för människovärdet.
8 § Mänskliga rättigheter och grundläggande friheter skall alltid beaktas vid etikprövningen samtidigt som hänsyn skall tas till intresset av att ny kunskap kan utvecklas genom forskning. Människors välfärd skall ges företräde framför samhällets och vetenskapens behov.
9 § Forskning får godkännas bara om de risker som den kan medföra för forskningspersoners hälsa, säkerhet och personliga integritet uppvägs av dess vetenskapliga värde.
10 § Forskning får inte godkännas, om det förväntade resultatet kan uppnås på ett annat sätt som innebär mindre risker för forskningspersoners hälsa, säkerhet och personliga integritet. Behandling av personuppgifter som avses i 3 § får godkännas bara om den är nödvändig för att forskningen skall kunna utföras.
11 § Forskning får godkännas bara om den skall utföras av eller under överinseende av en forskare som har den vetenskapliga kompetens som behövs.
11 a § /Upphör att gälla U:2004-05-01/ Vid etikprövning av klinisk undersökning på människor av ett läkemedels egenskaper (klinisk läkemedelsprövning) skall, utöver vad som följer av denna lag, bestämmelserna i 13 e och f §§ läkemedelslagen (1992:859) tillämpas.
Sverige har i tillegg personopplysningslovgivning, biobanklovgivning, legemiddellovgivning og andre reguleringer som mer eller mindre direkte kommer til anvendelse på medisinsk forskning.
19.4.2 Særlig om komitésystemet
Ny lov
Frem til 1. januar 2004 var det i Sverige forskningsetikkomiteer ved samtlige medisinske fakulteter. Det var ingen rettslig eller annen enhetlig regulering av komiteenes virksomhet med hensyn til organisering, sammensetning og arbeidsformer, og disse spørsmålene ble derfor løst noe ulikt. Komiteene var formelt sett rådgivende organer, men i praksis var forholdene omtrent som i Norge, dvs. at tilrådningene i praksis fungerte som tillatelser eller avslag (enkeltvedtak).
En departementsutredning om saken ble lagt frem i 2001 (Ds 2001:62 ”Etikprövning av forskning som avser människor”). På grunnlag av denne utredningen samt en høringsrunde, la regjeringen frem en proposisjon (2002/03:50) i januar 2003 om en ny lov om forskning på mennesker og biologisk materiale fra mennesker. Loven trådte i kraft 1. januar 2004.
Lovens begrunnelse
Begrunnelsen for lovforslaget var at både forsøkspersoners og forskeres rettsikkerhet trengte vern. Desssuten hadde de medisinske forskningsetiske komiteenes arbeid allerede karakter av myndighetsutøvelse, og de hadde i tillegg fått lovhjemlede oppgaver via den svenske personopplysningsloven. Slik virksomhet trengte forfatningsmessig støtte, og en regnet med at lovregulering av virksomheten ville skape bedre forutsetninger for en mer enhetlige vurderinger. Forskernes rettsikkerhet styrkes blant annet ved innføringen av en klageadgang. Videre er det vist til at en ratifisering av Oviedo-konvensjonen krever en rettslig regulering.
Hva loven gjelder
Det skal foretas en etisk vurdering dersom:
Forskningen innebærer et fysisk inngrep eller utføres med en metode som har til hensikt å påvirke mennesket fysisk eller psykisk.
Det skal drives forskning på biologisk materiale fra et levende eller dødt menneske, dersom materialet kan tilbakeføres til dette mennesket.
Forskningen skjer uten den enkeltes samtykke og det brukes følsomme personopplysninger. ”Følsomme personopplysninger” defineres som informasjon om rase eller etnisk opprinnelse, lovovertredelser, politiske oppfatninger, religiøs eller filosofisk overbevisning, medlemskap i fagforening, helse eller seksualliv er inkludert.
Komiteenes vurderinger
Loven angir en del prinsipper for den etiske vurderingen. Prinsippene er hovedsakelig hentet fra Oviedo-konvensjonen. Det er også gitt detaljerte bestemmelser om informasjon og samtykke. Forskningen må alltid utføres av eller være under overoppsyn av en forsker som har den nødvendige vitenskapelige kompetanse. Det er leder for et forskningsprosjekt som må søke om etisk vurdering. Godkjenning av et forskningsprosjekt har en varighet på to år.
Organisering av komitésystemet
Loven inneholder også regler om organisering av det forskningsetiske komitésystemet. Den etiske vurderingen av forskningsprosjekter skal utføres av seks regionale nemnder som skal være uavhengige forvaltningsorganer. En regional nemnd skal være inndelt i flere avdelinger som dekker ulike fagområder. Hver avdeling skal bestå av en leder og 15 øvrige medlemmer, som oppnevnes av regjeringen. Lederen må ha dommerkompetanse, og ti av de øvrige medlemmene må ha vitenskapelig kompetanse. Fem skal være lekrepresentanter eller representere ”allmenne interesser”.
Den sentrale nemnda, som også oppnevnes av regjeringen, skal bestå av en leder samt seks øvrige medlemmer. Fire av disse skal ha vitenskapelig kompetanse, mens to skal være lekrepresentanter eller representere ”allmenne interesser”. Også i denne nemnda må leder ha dommerkompetanse.
Den sentrale nemnden blir finansiert ved ”anslag”, dvs. midler over statsbudsjettet, mens de regionale nemndene blir finansiert ved hjelp av en avgift for hver søknad. Avgiften skal betales av prosjektleder.
Klage
Prosjektleder kan påklage beslutninger i en regional nemnd til en sentral nemnd. Det kan gjelde beslutninger der forskningen ikke er blitt godkjent som følge av den etiske vurderingen eller beslutninger der det har blitt satt vilkår som prosjektleder ikke kan godta. Den sentrale nemndas avgjørelse kan ikke påklages.
Ymse bestemmelser
Det finnes en straffebestemmelse, der den som med forsett utfører forskning uten å ha fått godkjenning eller ikke følger vilkår som har blitt stilt, kan straffes med bøter eller fengsel i høyst seks måneder. Regjeringen eller den myndighet regjeringen bestemmer kan fastsette nærmere forskrifter til loven.
20 Oppsummering – utvalgets vurdering av gjeldende reguleringer
20.1 Innledning
Utvalget er i mandatets punkt 2 bedt om å
vurdere sammenhengen mellom gjeldende reguleringer, herunder dagens bestemmelser om godkjenning fra ulike instanser, vurdering fra regional komité for medisinsk forskningsetikk, meldeordninger, samtykke og informasjon hjemlet i ulike lover innsyn mv. Det skal vurderes hvor tilgjengelig og oversiktlig gjeldende regler og retningslinjer er for forskere/forskningsinstitusjoner og for forsøkspersoner og givere av biologisk materiale til forskning.
Utvalget har gjennomgått gjeldende reguleringer, jf. kapittel 8-19 om gjeldende reguleringer med tilhørende tabeller. I tillegg har enkelte sentrale aktører kommet med synspunkter på gjeldende reguleringer. Utvalget har på denne bakgrunn reist spørsmål om medisinsk forskning er hensiktsmessig og klart regulert i dag.
I det følgende skal enkelte hovedproblemer med dagens reguleringer identifiseres. Utvalgets nærmere vurderinger av hva som er problematisk med gjeldende reguleringer og hva som er hensiktsmessig regulering, fremgår av utvalgets drøftelser av hvordan reguleringene bør være i del IV, V og VI.
20.2 Vurdering av gjeldende atferdsreguleringer
Som vist i kapittel 8-18 er medisinsk og helsefaglig forskningsvirksomhet i dag underlagt en rekke reguleringer. Utvalget har inntrykk av at medisinsk forskning i dag i all hovedsak drives forsvarlig. Utvalget er likevel av den oppfatning at dagens reguleringer ikke er optimale og vil i det følgende påpeke noen hovedproblemer med gjeldende atferdsreguleringer:
Fragmentert. Dagens regelverk er unødvendig fragmentert. Det vises her til oversikten i kapittel 10, herunder tabell 10.1. Ulike sider av medisinsk forskning er direkte eller indirekte regulert i over 40 forskjellige lover, forskrifter og ikke-juridiske retningslinjer (for eksempel profesjonsnormer). Det at regelverket er usedvanlig fragmentert, gjør at det er vanskelig å få oversikt. Dermed fremstår regelverket som utilgjengelig. Det er av den grunn vanskelig å vite hva som er gjeldende rett slik at for eksempel forskere og forskningsdeltakere, kan forutsi sin rettsstilling.
Uoversiktlig og utilgjengelig. Det at regelverket er fragmentert og dårlig harmonisert gjør at det er uoversiktlig og dermed vanskelig tilgjengelig for forskere, forsøkpersoner, myndigheter, kontroll- og tilsynsorganer og andre. Utvalget har konsultert en rekke forskere og myndighetsorganer, og det synes å være få, om noen, som har fullstendig oversikt over de til enhver tid gjeldende reguleringer av medisinsk og helsefaglig forskning.
Det er problematisk at få av de eksisterende reguleringer har som sitt primære formål å regulere (og/eller fremme) medisinsk og helsefaglig forskning, jf. kapittel 10. Dermed har enkelte reguleringer utilsiktede eller uheldige konsekvenser, ved at de vanskeliggjør forskning i unødvendig grad. Det er uheldig ikke bare sett fra forskerens, men også fra samfunnets side.
Dårlig harmoni. Medisinsk og helsefaglig forskning som involverer mennesker fordrer ofte bruk av humant biologisk materiale og helseopplysninger. I dag har man et relativt rigid og komplisert regelverk når det gjelder humant biologiske materiale og helseopplysninger, mens regelverket for forskning på mennesker er mangelfullt. Her er man stort sett henvist til å bruke alminnelige rettslige prinsipper og Helsinki-deklarasjonen som er vedtatt av Verdens legeforening, og som derfor i utgangspunktet bare gjelder for leger som selvpålagte profesjonsnormer.
Dårlig sammenheng. Det er videre ikke god sammenheng mellom de ulike reguleringene. Det kan synes som at ulike reguleringer er vedtatt uten at det er tatt tilstrekkelig hensyn til andre reguleringer. Det vises her til at det for eksempel er uklart i hvilken utstrekning pasientrettighetsloven, helsepersonelloven og bioteknologiloven kommer til anvendelse på forskning. Det vises også til kapittel 13 punkt kapittel 13.4 om individets rett til selvbestemmelse hvor det påvises at de ulike samtykkebestemmelsene ikke harmonerer helt. Herunder vises det til destruksjonsplikten i biobankloven som ikke samsvarer med legemiddelforskriftens krav om oppbevaring i 15 år. Oppbevaringsplikten er viktig av hensyn til tilsynsmyndighetenes mulighet til etterkontroll.
Mangler. Det er videre flere forhold ved medisinsk forskning som tilsynelatende ikke er regulert, eller – i beste fall – uklart regulert, men som burde vært klart regulert. Utvalget vil i denne sammenheng spesielt fremheve:
Uklare ansvarsforhold og ansvarsregler, jf. kapittel 16. Herunder foreligger det ingen generell plikt om å stille ansvarsforsikring ved forskning, og det er tvilsomt om de forsikringer som forskere (leger) har, dekker forskningsvirksomhet. Det hersker usikkerhet i forskningsmiljøene om i hvilken grad ordningen med Norsk Pasientskadeerstatning gjelder ved medisinsk og helsefaglig forskning.
Det eksisterer ikke noe klart forsvarlighetskrav til forskningsvirksomhet utover det som kan utledes fra alminnelige erstatningsrettslige prinsipper. Det må anses som uheldig. Det er uansett uklart hvilke krav som må være oppfylt for at forskningen skal kunne anses som forsvarlig, særlige gjelder dette forskning på mennesker.
Samtykkereglene er mangelfulle særlig når det gjelder forskning på barn, personer med redusert eller manglende samtykkekompetanse og forskning i kliniske nødsituasjoner.
Manglende krav til åpenhet og innsyn i forskningen.
Det er behov for en avklaring av forhold som knytter seg til kommersiell utnyttelse av og eiendomsrett til humant biologisk materiale og helseopplysninger som sådan. Økende grad av kommersialisering av forskning, betalers styring av forskningen og bindinger mellom finansieringskilder og forskere er også en utfordring. Økende kommersialisering av forskningen stiller store krav til hensiktsmessig og klar regulering. Utvalget har vanskeligheter med å se at gjeldende reguleringer er rustet til å møte denne utviklingen.
Alle forskere er ikke helsearbeidere, alle forskningsdeltakere er heller ikke pasienter. Det betyr at det lovverket som er laget for helseformål, verken omfatter forskere som ikke er helsearbeidere eller personer som ikke er pasienter. Det er særlig i slike tilfeller at gjeldende reguleringer synes mangelfulle.
Forholdet til internasjonal rett. Utvalget har også merket seg at mellom-statlige organisasjoner (særlig EU og Europarådet) har vedtatt og planlegger vedtatt reguleringer som (vil) stille(r) krav til reguleringen av medisinsk og helsefaglig forskning i Norge. Slik utvalget ser det, er ikke gjeldende reguleringer i overensstemmelse med verken gjeldende eller ventede folkerettslige forpliktelser. Særlig gjelder dette Oviedo-konvensjonen med tilleggsprotokoller.
20.3 Vurdering av kontroll- og tilsynssystemet
20.3.1 Generelt
Det vises til redegjørelsen for kontroll- og tilsynssystemet i kapittel 18. Det skilles i det følgende mellom forhåndskontroll, løpende tilsyn og etterhåndskontroll. I tillegg har utvalget vurdert etikkomitésystemet særskilt.
20.3.2 Forhåndskontroll
Uoversiktlig. Dagens system med forhåndskontroll er i likhet med selve regelverket, unødvendig fragmentert. Det fremstår dermed som uoversiktlig og unødig byråkratisk. Et forskningsprosjekt som berøres av mange reguleringer må forelegges en rekke ulike instanser, jf. kapittel 18 og figur 18.1. For eksempel vil en forsker kunne komme i den situasjon at hele eller deler av prosjektet må forelegges til godkjenning, tilrådning eller orientering for De regionale komiteer for forskningsetikk, Norsk Samfunnsvitenskapelig datatjeneste, Datatilsynet, Statens legemiddelverk, Sosial- og helsedirektoratet og Helse- og omsorgsdepartementet. Med så mange instanser involvert, kan det være vanskelig å vite hvem man skal henvende seg til. Her har imidlertid nevnte instanser tatt initiativ til å opprette en felles nettportal som skal gjøre det enklere for forskere å orientere seg.
Dobbeltbehandling. Et problem med dagens system er at flere instanser vurderer samme sider av et forskningsprosjekt. Dette er problematisk fordi det gjør prosessen unødvendig tidkrevende, byråkratisk og belastende for så vel forskeren som for myndighetene. Et annet problem er at enkelte sider av forskningsprosjektet vurderes av organer som ikke er sammensatt (kompetansemessig) med tanke på å vurdere de forhold som de vurderer. Det vises til at så godt som samtlige av de ovennevnte organer vurderer etiske spørsmål knyttet til for eksempel samtykke. Flere av dem vurderer studiets utforming og nytteverdi. Det finnes eksempler på at alle unntatt én instans har akseptert en strategi for innhenting av samtykke, med den følge at prosjektet dermed måtte stanses.
Uklar oppgavefordeling. Når ulike instanser har forskjellige (i verste fall motstridende) oppfatninger/råd, er det i enkelte tilfeller uklart hvem som egentlig har avgjørende myndighet/”siste ord”.
Særlig om REK. Det er problematisk at komitessystemet som har en så sentral plass og som i realiteten driver med myndighetsutøvelse, ikke er lovhjemlet. Det er videre prinsipielt uheldig at avgjørelsene ikke skal anses som enkeltvedtak, og at avgjørelsene ikke begrunnes eller er gjenstand for klage.
20.3.3 Løpende tilsyn og kontroll
Statens helsetilsyn fører tilsyn med helsetjenesten i Norge, men dette tilsynet omfatter i liten grad ikke-klinisk forskning. Helsetilsynet fører tilsyn etter biobankloven. Statens legemiddelverk fører tilsyn med klinisk utprøving av legemidler. Datatilsynet fører tilsyn med behandlingen av personopplysninger. Dermed er det klart at det ikke er etablert tilfredsstillende tilsynsordninger for deler av medisinsk og helsefaglig forskning. Særlig gjelder dette for forskning på mennesker.
20.3.4 Etterhåndskontroll
Dersom en ser bort fra domstolskontroll og hendelsesbasert tilsyn av Helsetilsynet, er det kun på området for klinisk utprøving av legemidler at det eksisterer et system for etterhåndkontroll gjennom en rapporteringsplikt. Denne ordningen fungerer imidlertid ikke tilfredsstillende.
Forskning handler om å opparbeide ny viten. Manglende rapportering av forskningsresultater (positive som negative) er derfor et problem. Man risikerer bl.a. at det forskes på problemstillinger det allerede er gjort urapporterte studier på, og med det utsettes kanskje enkeltpersoner for unødvendige inngrep.
20.4 Konklusjon
Utvalget mener at det på bakgrunn av de hovedproblemer som er beskrevet ovenfor og utvalgets kjennskap til dagens reguleringer for øvrig, utvilsomt er grunnlag for å si at dagens regulering av medisinsk og helsefaglig forskning ikke er hensiktsmessig.
I del IV, V og VI kommer derfor utvalget med anbefalinger til tiltak som skal sikre klar og hensiktsmessig regulering.
Fotnoter
Statutter for den internasjonale domstol, artikkel 38.
Oviedo-konvensjonen.
Porter (1997).
Porter (1997).
Beauchamp & Childress (2001).
Brody (1998).
Vogelsang (1968).
Blom (1973).
Vogelsang (1968).
Blom K (1973).
Blom K (1973).
Blom K (1973), Vogelsang (1968).
Blom K (1973).
NOU 2003: 33.
Ruyter (1997).
Annas & Grodin (1992).
Nürnberg-kodeksen.
Shuster (1998).
Møse (2003).
Ruyter (2000).
Jones (1993).
Ruyter (2003).
Beecher (1966).
Schartum & Bygrave (2004).
NOU 1993:22.
Selmer (1990).
Helsinki-deklarasjonen.
Se om disse kapittel 9 Utviklingstrekk
Ruyter et al (2000).
Vancouver-konvensjonen.
CIOMS-retningslinjer.
Etiske regler for leger.
Retningslinjer for samarbeid mellom legestand og farmasøytisk industri.
NENT (1992).
NOU 1999:13.
NEM (2001).
Aasen (2000) s 333-4, med videre henvisninger.
Stilltiende samtykke godtas i forhold til mindre inngripende og ikke risikofylt medisinsk behandling (se Aasen (2000) s. 353-6).
Schartum & Bygrave (2004) s. 96.
Schartum & Bygrave (2004) s. 96-97.
Oviedo-konvensjonen.
Oviedo-konvensjonens tilleggsprotokoll.
UNESCO (1997).
Bernt & Rasmussen (2003) s. 17- 23.
Bernt & Rasmussen (2003) s. 20.
Bernt & Rasmussen (2003) s. 24.
Punkt 8 i mandatet for de regionale komiteer for medisinsk forskningsetikk.
En noe mildere bestemmelse finnes i helsepersonelloven § 29, 1. ledd der det heter at ”departementet kan bestemme at opplysninger kan eller skal gis til bruk i forskning” uten hinder av taushetsplikt, men at det kan knyttes vilkår til et slikt vedtak. I slike situasjoner vil helsepersonellovens bestemmelser om taushetsplikt gjelde tilsvarende for forskeren.
Mandat for de regionale komiteer for medisinsk forskningsetikk, punkt 1.
Jf. Ot.prp. nr. 12 (1998-99) om lov om pasientrettigheter, hvor dette drøftes på s. 37-38, 82 og 126.
Med kloning menes teknikker for å fremstille arvemessig like kopier.
Innst. O. Nr. 58 (1998-99) s. 8.
Innst. O. Nr. 58 (1998-99) s. 7-8.
Syse (2001) s. 190.
Aasen (2000) s. 521-34.
Aasen (2000) s. 343-4.
Aasen (2000) s. 272.
Ot. Prp. Nr. 12 (1998-99) s. 132.
Aasen (2000) s. 347-52.
Ot. Prp. Nr. 12 (1998-99) s. 129.
Aasen (2000) s 453.
Ot. Prp. Nr 56 (2001-2002), datautskrift s. 40.
Ot. Prp. Nr 56 (2001-2002), datautskrift s. 40.
Ot. Prp. Nr 56 (2001-2002), datautskrift s. 40-41.
Se ovenfor i punkt 13.4.2.3.
Ot. Prp. Nr 56 (2001-2002), datautskrift s. 41.
Ot. Prp. Nr 56 (2001-2002), datautskrift s. 41.
Ot. Prp. Nr 56 (2001-2002), datautskrift s. 42.
Oviedo-konvensjonens tilleggsprotokoll.
Jf. for eksempel lov om statlig tilsyn med helsetjenesten, kommunehelsetjenesteloven, spesialisthelsetjenesteloven, tannhelsetjenesteloven, helsepersonelloven og pasientrettighetsloven.
Ot.prp. nr. 5 (1999 – 2000), merknadene til § 1.
Ot.prp. nr. 92 (1998-1999), side 101.
http://www.personvernnemnda.no
NOU 1993: 22.
Schartum & Bygrave (2004) s. 29 – 67.
Oviedo-konvensjonen.
Personopplysningsloven bruker uttrykket ”behandlingsansvarlig”, men innen helsesektoren vil ”behandlingsansvaret” lett kunne assosieres med den medisinske behandlingen. Derfor er ”data” satt først i ordet for å understreke hva behandlingen gjelder.
Nasjonalt folkehelseinstitutt er normalt behandlingsansvarlig for disse registrene.
Se definisjonen i helseregisterloven § 2 nr 2 og nærmere omtale nedenfor.
Se definisjonen i helseregisterloven § 2 nr 4 og nærmere omtale nedenfor.
Derimot ikke – nødvendigvis – selve identiteten, jf ”pseudonymisering”, straks nedenfor.
Enhver opplysning kan selvsagt krypteres, men her tar vi opp spørsmålet om kryptering av helseopplysninger.
Noe som trolig innebærer at manuell observasjon, bruk av medisinsk-teknisk apparatur, biokjemiske metoder, medisinske ekspertsystemer er blant de analysemetoder som kan være aktuelle.
Spørsmålet er imidlertid omstridt. Se Personvernnemndas avgjørelse nr 8/2002 som fastslår at humant materiale i seg selv ikke kan anses å være en ”personopplysning”.
Schartum & Bygrave (2004) skjelner mellom ”personobjekt” (jf biologisk materiale), persondata, personinformasjon og personopplysning for å beskrive kognitive og maskinelle prosesser som leder fra en fysisk person til en personopplysning, se s 108 – 111.
Det samme gjør forskriftene som regulerer sentrale helseregistre.
For sentrale helseregistre gjelder det egne forskriftsreguleringer som i de fleste tilfelle innebærer at mange helseopplysninger kan oppbevares i ”ubegrenset” tid.
Tilsvarende spørsmål er ikke løst etter personopplysningsloven, og kan der konkret by på stor tvil, for eksempel i forholdet mellom ungdom og foreldre.
Se personvernforskriften § 7 – 12.
Forskrift 2003.10.17 nr 1246
Forskrift 2002.06.21 nr 0567.
Forskrift 2003.06.20 nr 0740.
Forskrift 2001.12.21 nr 1476.
Forskrift 2001.12.21 nr 1477.
Forskrift 2001.12.21 nr 1483.
Forskrift 2003.06.20 nr 0739.
Forskrift 2003.11.14 nr 1353.
Tuberkulosekontrollforskriften avviker fra de øvrige forskriftene og den følgende beskrivelsen vil derfor ikke innbefatte denne forskriften.
Den enkelte forskrift regulerer også andre spørsmål i innledningskapittelet, men dette kommer vi ikke nærmere inn på her.
Formålsbestemmelsene i de sju forskriftene som har forskning som formål, finnes i § 1-3, og følger stort sett samme standardiserte formuleringsmåter.
For sammenstilling av opplysninger i Reseptregisteret, uttrykker vedkommende forskrift dette i klartekst i § 5-3 siste ledd.
Se for eksempel NORM-forskriften § 3-1 om utlevering av avidentifiserte opplysninger.
Stenvik (1999) s. 12.
Se Ot. prp. nr 67 (2001-2002).
Bernt & Rasmussen (2003).
www.hod.dep.no
www.shdir.no
Se Sosial- og helsedirektoratet, Skjema for søknad om fritak fra taushetsplikten i forbindelse med forskning: www.shdir.no/assets/3152/taushet3.doc
www.legemiddelverket.no
http://www.legemiddelverket.no/
Good Clinical Practice.
Sponsor: en person, et firma, en institusjon eller organisasjon som tar ansvaret for iverksetting, ledelse og/eller finansiering av en klinisk studie, og som undertegner søknaden, jf. forskriften § 1-2 bokstav g.
Sandaker et al (2004).
www.datatilsynet.no
www.helsetilsynet.no
Berg (1999).
Se Justisdepartementet, Høringsnotat – utkast til forskrift til forvaltningsloven, http://odin.dep.no/jd/norsk/publ/hoeringsnotater/012041-080076/index-dok000-b-n-a.html
www.bion.no
Se http://www.personvernnemnda.no/personvernnemnda.html
www.etikkom.no
Vancouver-konvensjonen.
http://www.etikkom.no/REK/SOP
http://www.nsd.uib.no/personvern/